 |
Composición:
Cada frasco-ampolla contiene: Paclitaxel (como nanopartķculas de Paclitaxel ligado a Albśmina) 100 mg. Luego de la reconstitución, cada ml de la suspensión contiene: 5 mg de Paclitaxel (como nanopartķculas de Paclitaxel ligadas a Albśmina). Excipientes: Solución de Albśmina Humana 700-1100 mg.
|
|
Descripción:
Abraxane®, un inhibidor de los microtśbulos, es una forma de paclitaxel unida a albśminas con un tamańo de partķcula promedio de aproximadamente 130 nanómetros. Paclitaxel existe en las partķculas en estado no cristalino y amorfo. Abraxane® se suministra como polvo blanco a amarillo, estéril, liofilizado para reconstitución con 20 ml de inyección de cloruro de sodio al 0.9%, USP antes de la infusión intravenosa. Cada vial descartable contiene 100 mg de paclitaxel (unido a albśmina humana) y aproximadamente 900 mg de albśmina humana (que contiene caprilato de sodio y acetiltriptofanato de sodio). Cada mililitro (ml) de suspensión reconstituida contiene 5 mg de paclitaxel. Abraxane® no contiene solventes. El agente activo de Abraxane® es paclitaxel. La denominación quķmica de paclitaxel es 5 · ,20-Epoxi-1,2 · ,4,7 · ,10 · ,13 · -hexahidroxitax-11-en-9-ona 4,10-dķacetato 2-benzoato 13- éster con (2R,3S)-N-benzoil-3-fenilisoserina. Paclitaxel es un polvo cristalino blanco a blanquecino cuya formula empķrica es C47H51NO14 y su peso molecular es 853,91. Es altamente lipofķlico, insoluble en agua y se funde a aproximadamente 216°C a 217°C.
|
|
Indicaciones:
Cįncer de mama metastįsico: Abraxane® estį indicado para tratar el cįncer de mama en caso de fracasar la quimioterapia combinada para la enfermedad metastįsica o la recidiva dentro de los 6 meses de quimioterapia adyuvante. El tratamiento previo debe haber incluido una antraciclina, a menos que esta esté clķnicamente contraindicada. Cįncer de pulmón no microcķtico: Abraxane® estį indicado para el tratamiento de primera lķnea de cįncer de pulmón no microcķtico localmente avanzado o metastįsico en combinación con carboplatino en pacientes que no son candidatos para cirugķa curativa o terapia de radiación. Adenocarcinoma del pįncreas: Abraxane® estį indicado para el tratamiento de primera lķnea de pacientes con adenocarcinoma metastįsico del pįncreas, en combinación con gemcitabina.
|
|
Propiedades:
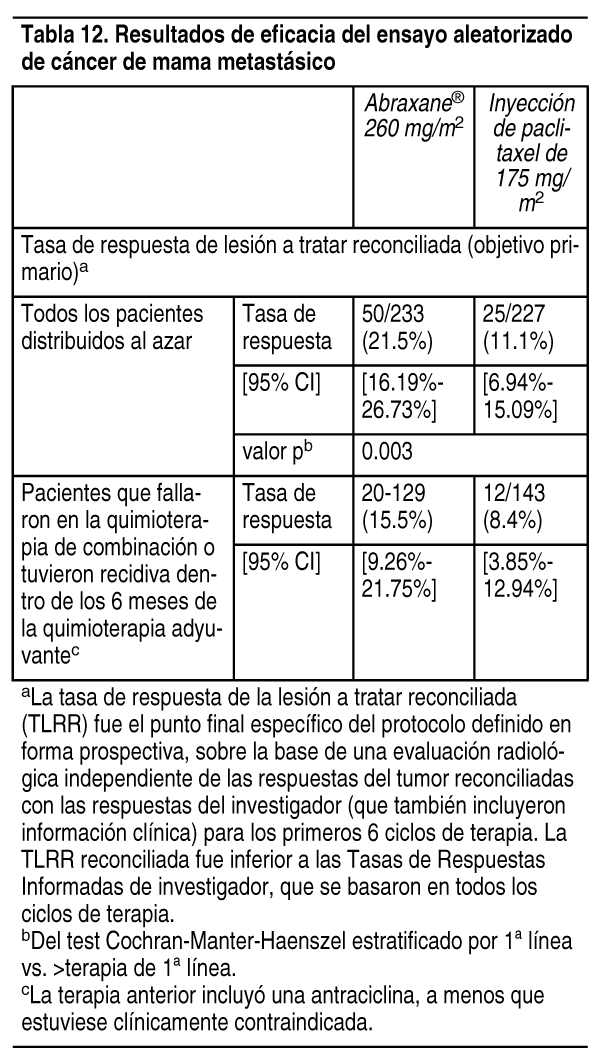
Farmacologķa clķnica: Mecanismo de acción: Abraxane® es un inhibidor de microtśbulos que estimula el ensamblado de microtśbulos a partir de los dķmeros de tubulina y estabiliza los microtśbulos impidiendo su despolimerización. Esta estabilización inhibe la dinįmica de reorganización normal de la red de microtśbulos, esencial para las funciones celulares vitales en las fases mitótica e interfase. Paclitaxel induce la formación de grupos anormales o "haces" de microtśbulos a lo largo de todo el ciclo celular y de husos mśltiples de microtśbulos durante la mitosis. Farmacocinética: Absorción: Estudios clķnicos determinaron la farmacocinética de paclitaxel total tras perfusiones de 30 a 180 minutos de Abraxane® con niveles de dosis de 80 a 375 mg/m2. Los niveles de dosis de mg/m2 se refieren a mg de paclitaxel en Abraxane®. Tras la administración intravenosa de Abraxane®, las concentraciones plasmįticas de paclitaxel disminuyeron en forma bifįsica; la disminución rįpida inicial representa la distribución al compartimiento periférico y la segunda fase mįs lenta representa la eliminación de la droga. La exposición a la droga (AUCs) fue proporcional a la dosis de 80 a 375 mg/m2 y la farmacocinética de paclitaxel fue independiente de la duración de la administración intravenosa de Abraxane®. Los datos farmacocinéticos de 260 mg/m2 de Abraxane® administrado durante una infusión de 30 minutos se compararon con la farmacocinética de 175 mg/m2 de inyección de paclitaxel durante una infusión de 3 horas. El clearance fue mayor (43%) y el volumen de distribución de Abraxane® también fue mayor (53%) al de la inyección de paclitaxel. No hubo diferencias en las vidas medias terminales. Distribución: Luego de la administración de Abraxane a pacientes con tumores sólidos, el paclitaxel se distribuye uniformemente en las células sanguķneas y en el plasma y se une significativamente a las proteķnas plasmįticas (94%). En un estudio comparativo intrapaciente, la fracción de paclitaxel no ligado en el plasma fue significativamente mayor con Abraxane (6.2%) que con paclitaxel basado en solventes (2.3%). Esto contribuye a una exposición significativamente mayor al paclitaxel no ligado con Abraxane en comparación con paclitaxel basado en solventes, donde la exposición total es comparable. Los estudios in vitro de unión a las proteķnas séricas humanas usando concentraciones de paclitaxel que variaron de 0.1 a 50 µg/ml indicaron que la presencia de cimetidina, ranitidina, dexametasona o difenhidramina no afectaron la unión a proteķnas del paclitaxel. El volumen de distribución total es de aproximadamente 1741 I; el gran volumen de distribución indica una extensa distribución extravascular y/o unión tisular del paclitaxel. Metabolismo: Estudios in vitro con microsomas de hķgado humano y preparados de tejidos mostraron que paclitaxel fue metabolizado principalmente a 6 a -hidroxipaclitaxel por CYP2C8; y a 2 metabolitos menores, 3'-p-hidroxipaclitaxel y 6 a , 3'-p-dihidroxipaclitaxel, por CYP3A4. In vitro, el metabolismo de paclitaxel a 6 a -hydroxypaclitaxel fue inhibido por una cantidad de agentes (ketoconazol, verapamilo, diazepam, quinidina, dexametasona, ciclosporina, teniposido, etoposido y vincristina), pero las concentraciones usadas excedieron las encontradas in vivo tras dosis terapéuticas normales. La testosterona, el 17 a -etinil estradiol, el įcido retinoico y quercetina, un inhibidor especifico de CYP2C8, también inhibieron la formación de 6 a - hidroxipaclitaxel in vitro. La farmacocinética de paclitaxel también puede modificarse in vivo como consecuencia de las interacciones con compuestos que son medios, inductores o inhibidores de CYP2C8 y/o CYP3A4 (ver Interacciones medicamentosas). Eliminación: En el rango de dosis clķnica de 80 a 300 mg/m2, el aclaramiento total promedio de paclitaxel varķa entre 13 y 30 I/h/m2 y la vida media terminal promedio varķa entre 13 y 27 horas. Tras una infusión de 30 minutos de dosis de 260 mg/m2 de Abraxane®, los valores medios de la recuperación urinaria acumulativa de la droga no metabolizada (4%) indicaron una amplia clearance no renal. Menos del 1% de la dosis total administrada se excretó en la orina como los metabolitos 6 a -hidroxipaclitaxel y 3'-p-hidroxipaclitaxel. La excreción fecal fue aproximadamente el 20% de la dosis total administrada. Poblaciones especķficas: Farmacocinética en la insuficiencia hepįtica: El efecto de la insuficiencia hepįtica sobre la farmacocinética de paclitaxel luego de la administración de Abraxane se estudió en pacientes con tumos res sólidos avanzados. Los resultados mostraron que la insuficiencia hepįtica leve (bilirrubina total >1 a £ 1.5 x ULN, AST £ 10 x ULN, n=8) no tuvo efectos clķnicamente importantes sobre la farmacocinética de paclitaxel. Los pacientes con insuficiencia hepįtica moderada (bilirrubina total >1.5 a £ 3 x ULN, AST £ 10 x ULN, n=5) tuvieron una disminución de la tasa de eliminación de paclitaxel del 22% al 26% y un aumento aproximado del 20% en el AUC promedio de paclitaxel en comparación con los pacientes con función hepįtica normal (bilirrubina total £ ULN, AST £ ULN, n=130 (ver Posologķa y Administración y Uso en poblaciones especķficas). La eliminación de paclitaxel muestra una correlación inversa con la bilirrubina total y una correlación positiva con la albśmina sérica. Los modelos farmacocinéticos/farmacodinįmicos indican que no existe correlación entre la función hepįtica (segśn lo indicado por la albśmina basal o por el nivel de bilirrubina total) y la neutropenia luego de los ajustes por exposición a Abraxane. No se dispone de datos farmacocinéticos para pacientes con bilirrubina total > x ULN o para los pacientes con adenocarcinoma metastįsico del pįncreas (ver Posologķa y administración , y Uso de poblaciones especķficas). Farmacocinética en la insuficiencia renal: El efecto de la insuficiencia renal preexistente leve (aclaramiento de creatinina ³ 60 a <90 ml/min, n=61) o moderada (aclaramiento de creatinina ³ 30 a <60 ml/min, n=23) sobre la farmacocinética de paclitaxel luego de la administración de Abraxane se estudió en pacientes con tumores sólidos avanzados. La insuficiencia renal leve a moderada no tuvo efectos clķnicamente importantes sobre la tasa de eliminación mįxima y la exposición sistémica (AUC y Cmax) de paclitaxel (ver Uso en poblaciones especķficas). Otros factores intrķnsecos: Los anįlisis farmacocinéticos poblacionales de Abraxane muestran que el peso corporal (40 a 143 kg), el įrea de superficie corporal (1.3 a 2.4 m2), el sexo, la raza (asiįtica vs. blanca), la edad (24 a 85 ańos) y el tipo de tumor sólido no tienen un efecto clķnicamente importante sobre la tasa de eliminación mįxima y la exposición sistémica (AUCX y Cmax) de paclitaxel. Interacciones farmacocinéticas entre carboplatino y Abraxane®: La administración de carboplatino inmediatamente después de completar la infusión de Abraxane® en pacientes con cįncer de pulmón no microcķtico no causó cambios clķnicamente importantes en la exposición a paclitaxel. La media de AUCinf observada de carboplatino libre fue aproximadamente un 23% mįs alta que el valor a alcanzar (6 min*mg/ml) pero su vida media y el clearance fueron consistentes con los reportados en la ausencia de paclitaxel. Interacciones farmacocinéticas entre Abraxane® y gemcitabina: No se han estudiado las interacciones farmacocinéticas entre Abraxane® y gemcitabina en humanos. Toxicologķa no clķnica: Carcinogénesis, mutagénesis, trastornos de la fertilidad: No se ha estudiado el potencial carcinogénico de Abraxane®. Paclitaxel ha demostrado ser clastogenico in vitro (induce aberraciones cromosómicas en linfocitos humanos) e in vivo (test de micronścleos en ratones). Abraxane® no produjo mutagenicidad en el test de Ames o en el ensayo de mutación génica CHO/HGPRT. La administración a ratas macho de 42 mg/m2 de partķculas de paclitaxel unidas a proteķnas sobre una base semanal (aproximadamente el 16% de la exposición mįxima diaria recomendada para humanos sobre la base de la superficie corporal) por 11 semanas antes del apareamiento con ratas hembra no tratadas produjo una reducción significativa de la fertilidad, acompańada por una disminución de las tasas de preńez y un aumento de la pérdida de embriones en las hembras apareadas. También se observó una baja incidencia de anormalidades fetales esqueléticas y del tejido blando con dosis de 3 y 12 mg/m2/semana en este estudio (aproximadamente del 1 al 5% de la exposición mįxima diaria recomendada para humanos sobre la base de mg/m2). Se observó atrofia/degeneración testicular en estudios toxicológicos de dosis śnica en roedores a los que se les administraron partķculas de paclitaxel unidas a proteķnas con dosis inferiores a la dosis recomendada para humanos; las dosis fueron de 54 mg/m2 en roedores y 175 mg/m2 en perros. Estudios clķnicos: Cįncer de mama metastįsico: Datos procedentes de 106 pacientes reclutados en 2 ensayos clķnicos abiertos no controlados y de 460 pacientes inscriptos en un ensayo comparativo y aleatorio avalan el uso de Abraxane® en el tratamiento del cįncer de mama metastįsico. Ensayos abiertos de un solo grupo: En un ensayo, se administró una dosis de 175 mg/m2 de Abraxane® en infusión durante 30 minutos a 43 pacientes con cįncer de mama metastįsico. El segundo ensayo utilizó una dosis de 300 mg/m2 en infusión de 30 minutos en 63 pacientes con cįncer de mama metastįsico. Los ciclos se administraron con intervalos de 3 semanas. Se observaron respuestas objetivas en ambos estudios. Ensayo comparativo y aleatorio: Este ensayo multicéntrico se realizó en 460 pacientes con cįncer de mama metastįsico, que fueron distribuidos al azar para recibir Abraxane® a una dosis de 260 mg/m2 dada como infusión de 30 minutos o una inyección de paclitaxel de 175 mg/m2 dada como infusión de 3 horas. El 64% de los pacientes tenķa una alteración del estado funcional (ECOG 1 ó 2) al comienzo del ensayo; el 79% tenķa metįstasis viscerales; y el 76% presentaba > 3 localizaciones metastįsicas. El 14% de los pacientes no habķa recibido quimioterapia previa, el 27% habķa recibido quimioterapia solo durante el tratamiento adyuvante, el 40% como tratamiento de la enfermedad metastįsicas y el 19% como tratamiento adyuvante y metastįsico. El 59% de los pacientes recibió el medicamento de estudio como terapia en segunda lķnea o posteriores. El 77% de los pacientes habķa recibido tratamiento previo con antraciclinas. En este ensayo, los pacientes de la rama de tratamiento con Abraxane® tenķan una tasa de respuesta de la lesión a tratar reconciliada estadķstica y significativamente mayor (el punto final primario del ensayo) del 21.5% (95% CI: del 16.2% al 26.7%), comparado con el 11.1% (95% CI: del 6.9% al 15.1%) para los pacientes de la rama de tratamiento con inyección de paclitaxel. No hubo una diferencia estadķsticamente significativa en la supervivencia general entre las 2 ramas del estudio.
Ver Tabla
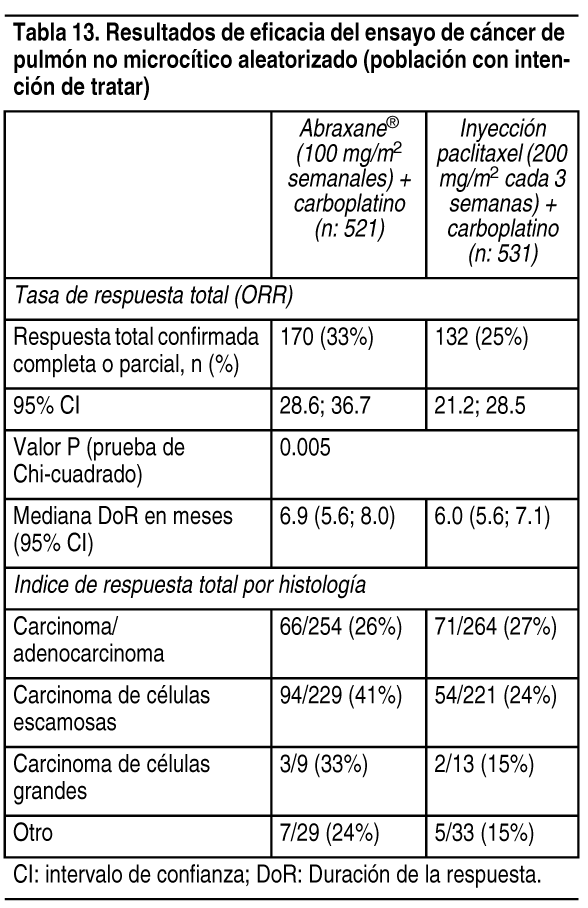
Cįncer de pulmón no microcķtico: Se realizó un estudio abierto, multicéntrico, aleatorizado en 1052 pacientes sin tratamiento quimioterįpicos previo con cįncer de pulmón no microcķtico en etapa IIIb/IV para comparar Abraxane® en combinación con carboplatino con inyección de paclitaxel en combinación con carboplatino como tratamiento de primera lķnea en pacientes con cįncer de pulmón no microcķtico avanzado. Abraxane® fue administrado como infusión intravenosa durante 30 minutos a una dosis de 100 mg/m2 en los dķas 1, 8 y 15 de cada ciclo de 21 dķas. La inyección de paclitaxel fue administrada como una infusión intravenosa durante 3 horas a una dosis de 200 mg/m2, luego de la premeditación. En ambos grupos, de tratamiento, carboplatino fue administrado a una dosis de AUC = 6 mg · min/ml de forma intravenosa en el dķa 1 de cada ciclo 21 dķas luego de completar de Abraxane®/infusión de paclitaxel. El tratamiento fue administrado hasta la progresión de la enfermedad o el desarrollo de una toxicidad no aceptable. La medida de resultado de eficacia primaria fue la tasa de respuesta total tal como determino el comité central de revisión independiente por medio de las normativas RECIST (Versión 1.0). En la población con intención de tratar (todos aleatorizados), la mediana de edad era de 60 ańos, el 75%eran hombres, el 81% eran de raza blanca, el 49% tenķan un adenocarcinoma, el 43% tenķan cįncer de pulmón de células escamosas, el 76% tenķa la escala ECOG PS 1, y el 73% fumaba o habķa fumado. Los pacientes recibieron una mediana de 6 ciclos de tratamiento en ambos grupos de estudio. Los pacientes en el grupo Abraxane®/carboplatino tuvieron una mayor tasa de respuesta total estadķsticamente significativa en comparación con pacientes en el grupo de inyección de paclitaxel/carboplatino (33% versus 25%). No habķa una diferencia estadķsticamente significativa en la sobrevida total entre los 2 grupos de estudio.
Ver Tabla
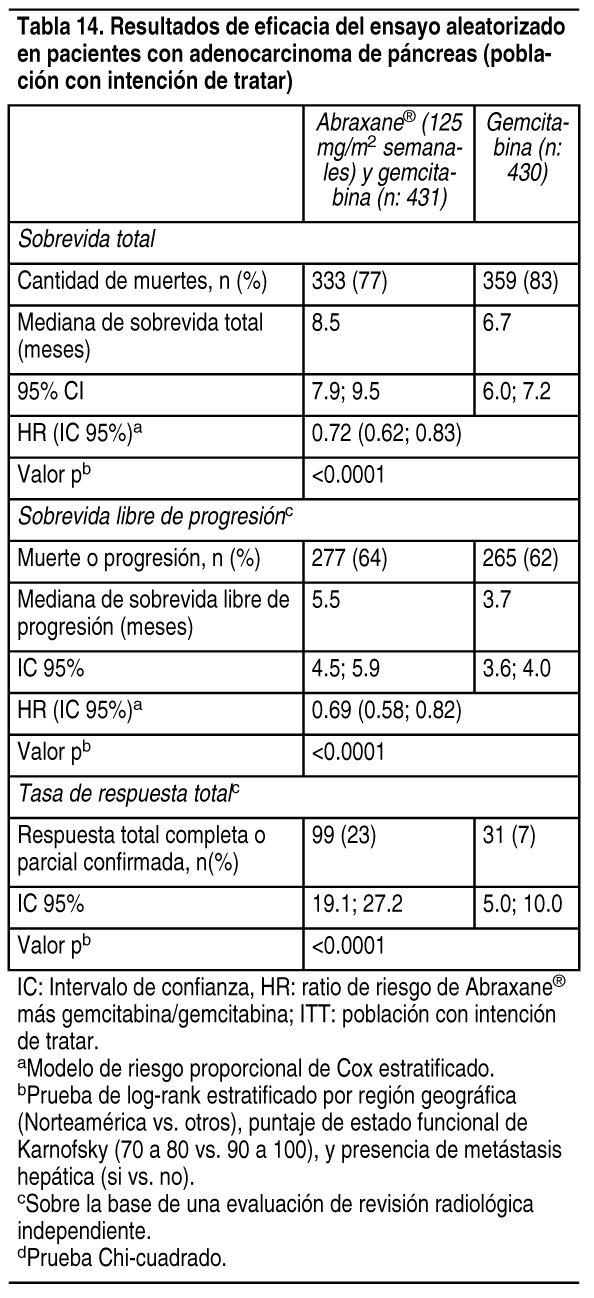
Adenocarcinoma del pįncreas: Se realizó un estudio abierto, randomizado, multinacional y multicéntrico en 861 pacientes que comparó Abraxane® mįs gemcitabina frente a monoterapia de gemcitabina como tratamiento de primera lķnea del adenocarcinoma metastįsico del pįncreas. Los criterios clave de elegibilidad fueron estado funcional de Karnofsky (KPS) ³ 70, nivel normal de bilirrubina, niveles de transaminasa £ 2.5 veces el lķmite superior normal (ULN) o £ 5 veces el ULN para pacientes con metįstasis del hķgado, sin quimioterapia citotóxica previa en el escenario adyuvante o para enfermedad metastįsica, sin infección activa en curso que requiera terapia sistémica, y sin antecedentes de enfermedad pulmonar intersticial. Los pacientes con una rįpida disminución del KPS ( ³ 10%) o albśmina sérica ( ³ 20%) durante el perķodo de selección de 14 dķas previo a la randomización del estudio no fueron elegibles. Se randomizaron 861 pacientes (1:1) en la rama Abraxane®/gemcitabina (N = 431) o en la rama gemcitabina (N = 430). La randomización se estratificó por región geogrįfica (Australia, Europa Occidental, Europa del Este o Norteamérica), KPS (70 a 80 vs. 90 a 100), y la presencia de metįstasis hepįticas (sķ vs. No). Los pacientes randomizados en Abraxane®/gemcitabina recibieron Abraxane® 125 mg/m² como infusión intravenosa durante 30 - 40 minutos seguido de gemcitabina 1000 mg/m² como infusión intravenosa durante 30 - 40 minutos en los Dķas 1, 8 y 15 de cada ciclo de 28 dķas. Los pacientes ramdomizados en gemcitabina recibieron 1000 mg/m² como infusión intravenosa durante 30-40 minutos semanalmente por 7 semanas seguido de un perķodo de descanso de 1 semana en el Ciclo 1, luego como 1000 mg/m² en los Dķas 1, 8 y 15 de cada ciclo subsiguiente de 28 dķas. Los pacientes de ambas ramas recibieron tratamiento hasta la progresión de la enfermedad o toxicidad no aceptable. La principal medición de resultados de eficacia fue la sobrevida total (OS). Otras mediciones de resultados fueron la sobrevida libre de progresión (PFS) y la tasa de respuesta total (ORR), ambas evaluadas mediante la revisión radiológica cegada central independiente usando RECIST (versión 1.0). En la población con intención de tratar (todos randomizados), la mediana de edad fue de 63 ańos (rango 27 - 88 ańos) con 42% ³ 65 ańos; el 58% eran hombres; el 93% eran de raza blanca y el KPS era de 90-100 en el 60%. Las caracterķsticas de la enfermedad incluyeron 46% de pacientes con 3 o mįs sitios metastįsicos; 84% de los pacientes tenķa metįstasis del hķgado; y la ubicación de la lesión pancreįtica primaria fue en la cabeza (43%), en el cuerpo (31%) o en la cola (25%) del pįncreas. En la tabla 14 se muestran los resultados de sobrevida total, sobrevida libre de progresión y tasa de respuesta total.
Ver Tabla
En anįlisis exploratorios realizados en subgrupos clķnicamente relevantes con una cantidad suficiente de individuos, los efectos del tratamiento en la sobrevida total fueron similares a aquellos observados en la población total del estudio.
|
|
Posologķa:
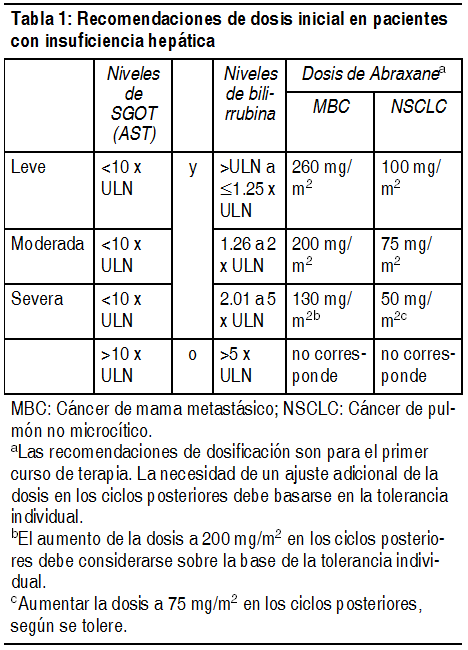
Cįncer de mama metastįsico: Tras fracasar la quimioterapia combinada para el cįncer de mama metastįsico o la recidiva dentro de los 6 meses de quimioterapia adyuvante, la dosis recomendada de Abraxane® es 260 mg/m2 administrados en forma intravenosa sobre 30 minutos cada 3 semanas. Cįncer de pulmón no microcķtico: La dosis recomendada de Abraxane® es 100 mg/m2 administrada como infusión intravenosa durante 30 minutos en los dķas 1, 8 y 15 de cada ciclo de 21 dķas. Administrar gemcitabina inmediatamente después de Abraxane® (ver Estudios clķnicos). Adenocarcinoma del pįncreas: La dosis recomendada de Abraxane® es 125 mg/m2 administrada como infusión intravenosa durante 30-40 minutos en los dķas 1, 8 y 15 de cada ciclo de 28 dķas. Administrar gemcitabina inmediatamente después de Abraxane® en los dķas 1,8 y 15 de cada ciclo de 28 dķas (ver Estudios clķnicos). Dosificación en pacientes con insuficiencia hepįtica: No es necesario ajustar la dosis en pacientes con insuficiencia hepįtica leve. Los pacientes con insuficiencia hepįtica moderada y severa tratados con Abraxane® pueden tener un riesgo mayor de toxicidades conocidas al paclitaxel. Suspender Abraxane® si la AST > 10 x ULN o la bilirrubina > 5.0 x ULN. La Tabla 1 muestra las recomendaciones de ajuste de la dosis para el primer curso de terapia. En cįncer de mama metastįsico, la dosis de Abraxane® puede aumentarse de 130 mg/m2 hasta 200 mg/m2 en pacientes con insuficiencia hepįtica severa en los ciclos posteriores, segśn la tabla 1. En cįncer de pulmón no microcķtico, reducir la dosis de Abraxane® a 50 mg/m2 en pacientes con insuficiencia hepįtica severa. En ciclos posteriores, la dosis de Abraxane® podrķa incrementarse a 75 mg/m2, segśn se tolere. Los pacientes deben tener un control estricto.
Ver Tabla
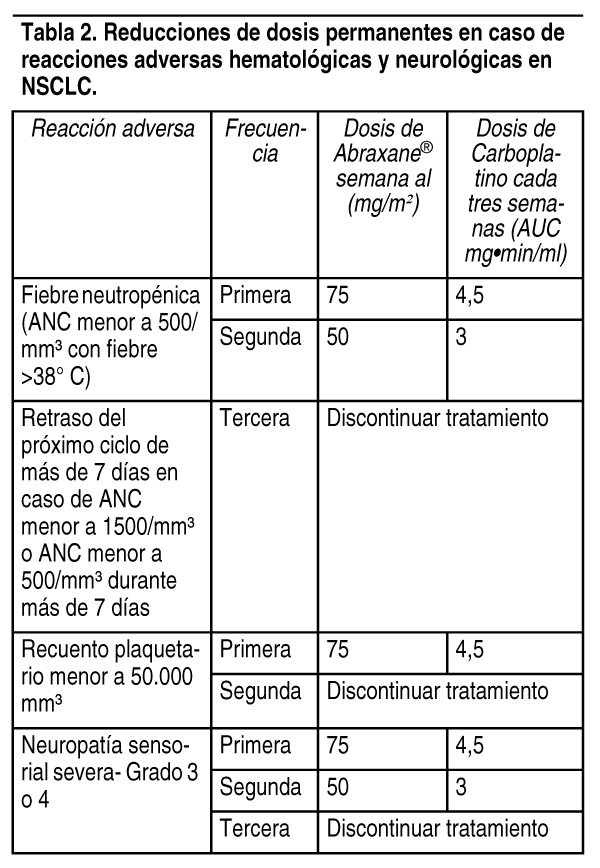
Recomendaciones de reducción de la dosis/discontinuación: Cįncer de mama metastįsico: En los pacientes que presentan neutropenia severa (neutrófilos <500 células/mm3 durante 1 semana o mįs) o neuropatķa sensitiva severa durante el tratamiento con Abraxane®, debe reducirse la dosis a 220 mg/m2 en los ciclos posteriores de Abraxane®. En caso de recurrencia de neutropenia severa o de neuropatķa sensitiva severa, efectuar una nueva reducción de la dosis a 180 mg/m2. En caso de neuropatķa sensitiva de Grado 3, interrumpir el tratamiento hasta la resolución a Grado 1 ó 2 y a continuación reducir la dosis para todos los ciclos posteriores de Abraxane® (ver Contraindicaciones, Advertencias y Precauciones y Efectos colaterales). Cįncer de pulmón no microcķtico: No administrar Abraxane® en el dķa 1 del ciclo hasta que el recuento absoluto de neutrófilos (ANC) sea al menos de 1.500 células/mm3 y el recuento plaquetario sea al menos de 100.000 células/mm3 (ver Contraindicaciones, Advertencias y Precauciones). En pacientes que desarrollan trombocitopenia o neutropenia severa suspender el tratamiento hasta que los recuentos se recuperen a un recuento absoluto de neutrófilos de al menos 1.500 células/mm3 y un recuento de plaquetas de al menos 100.000 células/mm3, en el dķa 1 o un recuento absoluto de neutrófilos de al menos 500 células/mm3 y un recuento de plaquetas de al menos 50.000 células/mm3 en los dķas 8 ó 15 del ciclo. Al reiniciar la dosis, reducir permanentemente las dosis de Abraxane® y carboplatino tal como figura en la tabla 2. Suspender Abraxane® en caso de neuropatķa periférica de Grado 3-4. Retomar Abraxane® y carboplatino en dosis reducidas (ver Tabla 2) cuando la neuropatķa periférica mejore a Grado 1 o se haya resuelto por completo (ver Advertencias y Precauciones y Efectos colaterales).
Ver Tabla
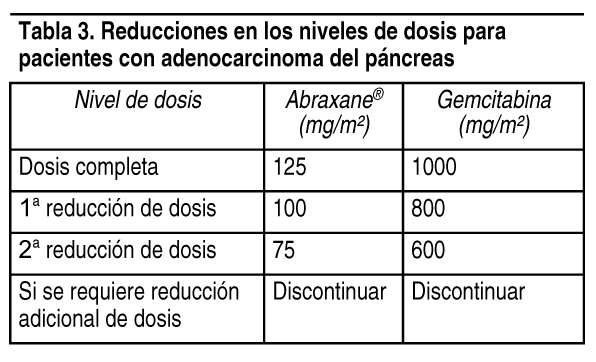
Adenocarcinoma del pįncreas: En la Tabla 3 se suministran las reducciones de los niveles de dosis para pacientes con adenocarcinoma del pįncreas, segśn se hace referencia en las Tablas 4 y 5.
Ver Tabla
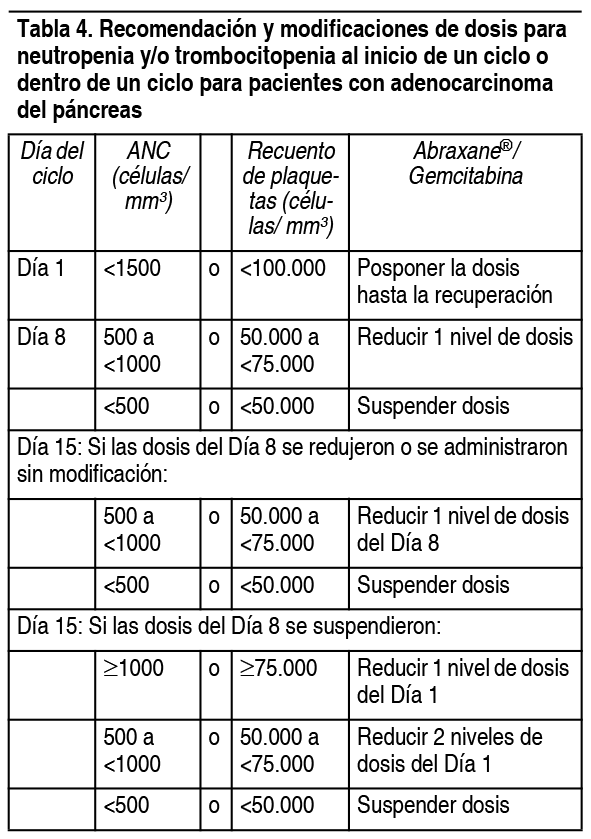
En la Tabla 4 se brindan las modificaciones de la dosis recomendadas para neutropenia y trombocitopenia para pacientes con adenocarcinoma del pįncreas.
Ver Tabla
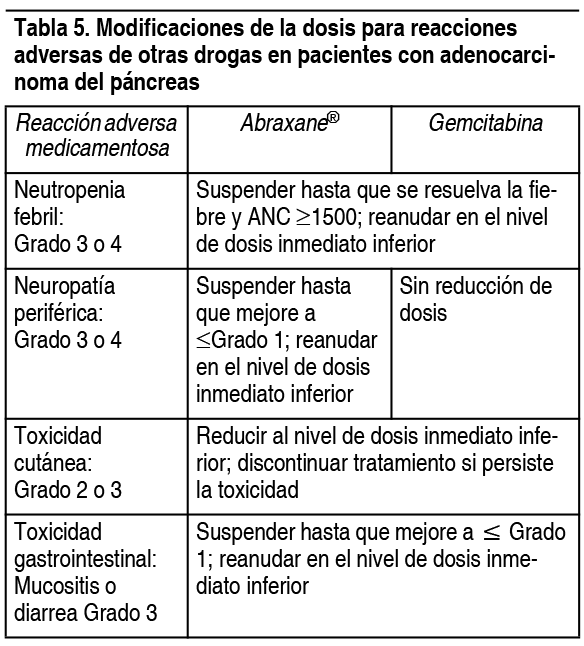
En la tabla 5 se brindan las modificaciones de dosis recomendadas para otras reacciones adversas medicamentosas en pacientes con adenocarcinoma del pįncreas.
Ver Tabla
Precauciones de preparación y administración: Abraxane® es un fįrmaco citotóxico y por lo tanto debe manipularse con precaución, al igual que ocurre con otros compuestos de paclitaxel potencialmente tóxicos. Se recomienda usar guantes. Si Abraxane® (polvo liofilizado o suspensión reconstituida) entra en contacto con la piel, esta debe lavarse inmediatamente y a fondo con jabón y agua. Tras la exposición tópica a paclitaxel, las reacciones pueden incluir picazón, ardor y rojez. Si Abraxane® entra en contacto con las membranas mucosas, estas deben lavarse a fondo con agua abundante. Debido a la posibilidad de extravasación, es aconsejable monitorear de cerca el lugar de infusión por si esta se produce durante la administración del medicamento. Limitando el tiempo de infusión de Abraxane® a 30 minutos, de acuerdo a las instrucciones, se reduce la probabilidad de reacciones asociadas a la infusión (ver Efectos colaterales). No es necesaria una medicación previa para evitar las reacciones de hipersensibilidad antes de la administración de Abraxane®. Puede requerirse medicación previa en pacientes con antecedentes de reacciones de hipersensibilidad al Abraxane®. En los pacientes que hayan presentado una reacción de hipersensibilidad severa al Abraxane® no deberķa reiniciarse la terapia con esta droga (ver Advertencias y Precauciones). Preparación para administración intravenosa: Abraxane® se presenta como polvo liofilizado estéril para su reconstitución antes de su uso. Para evitar errores, leer todas las instrucciones de preparación antes de la reconstitución. 1) En forma aséptica, reconstituir cada vial inyectando 20 ml de solución de cloruro de sodio al 0.9%, USP. 2) Inyectar lentamente los 20 ml de solución de cloruro de sodio al 0.9%, USP, durante por lo menos 1 minuto, usando una jeringa estéril para dirigir el flujo de la solución hacia el interior de la pared del vial.
Ver Tabla
3) No inyectar la solución de cloruro de sodio al 0.9%, USP, directamente sobre el polvo liofilizado ya que se producirį espuma. 4) Una vez que se haya completado la inyección, dejar reposar el vial durante por lo menos 5 minutos para asegurar la humectación adecuada del polvo liofilizado. 5) Agitar suavemente y/o invertir el vial lentamente durante por lo menos 2 minutos hasta completar la disolución del polvo. Evitar la formación de espuma. 6) Si se forma espuma o grumos, se debe dejar reposar la solución durante por lo menos 15 minutos hasta que desaparezca la espuma. Cada ml de fórmula reconstituida contiene 5 mg/ml de paclitaxel (como nanopartķculas de paclitaxel ligado a albśmina). La suspensión reconstituida debe tener un aspecto lechoso y homogéneo sin precipitados visibles. Si se observa precipitación o sedimentación, se debe invertir de nuevo el vial suavemente para asegurar la re-suspensión completa antes de su uso. Descartar la suspensión reconstituida si se observan precipitados. Descartar cualquier parte no usada. Calcular el volumen de dosis total exacta de suspensión de 5 mg/ml necesaria para el paciente y extraer lentamente el volumen de dosis de la suspensión reconstituida de la(s) ampolla(s) e introducirlo en la jeringa: Volumen de dosis (ml) = Dosis total (mg)/5 (mg/ml). Inyectar la cantidad apropiada de Abraxane® reconstituido en una bolsa de infusión IV vacķa, estéril [recipientes de cloruro de polivinilo (PVC) con plasticidad, bolsa IV de tipo PVC o no PVC]. No es necesario el uso de envases de solución especiales sin DEHP o de sets de administración para preparar o administrar perfusiones de Abraxane®. El uso de dispositivos médicos que contengan aceite siliconado como lubricante (es decir, jeringas y bolsas intravenosas) para reconstituir y administrar Abraxane puede resultar en la formación de hebras proteinįceas. Inspeccionar visualmente la suspensión reconstituida de Abraxane que esta dentro de la bolsa intravenosa antes de su administración. Desechar la suspensión reconstituida si se observan hebras proteinįceas, particular o cambios de coloración. Estabilidad: Los viales sin abrir de Abraxane® permanecen estables hasta la fecha indicada en el envase mientras se conserven a temperatura no mayor a 30°C en el envase original. El congelamiento o refrigeración no afectan negativamente la estabilidad del producto. Estabilidad de la suspensión reconstituida en el vial: Abraxane® reconstituido en el vial debe usarse inmediatamente, pero puede mantenerse de 2°C a 8°C (36°F a 46°F) por un mįximo de 8 horas, en caso de ser necesario. Si no se usa inmediatamente, cada vial de suspensión reconstituida debe colocarse nuevamente en el estuche original para protegerlo de la luz intensa. Descartar cualquier parte no usada. Estabilidad de la suspensión reconstituida en la bolsa de infusión: La suspensión para infusión, cuando se prepara segśn las recomendaciones de la bolsa de infusión, debe usarse en forma inmediata. Refrigerarse entre 2° C a 8° C (36°F a 46°) y protegerse de la luz intensa por un mįximo de 24 horas. El tiempo total combinado de almacenamiento refrigerado de Abraxane reconstituido dentro del vial y dentro de la bolsa de infusión es de 24 horas. Esto puede seguirse por un almacenamiento dentro de la bolsa de infusión a temperatura ambiente (aproximadamente 25° C) y expuesto a la luz por un mįximo de 4 horas. Descartar cualquier parte no usada. Formas de dosificación y concentraciones: Para la suspensión inyectable: Polvo liofilizado con 100 mg de paclitaxel formulado como nanopartķculas de paclitaxel ligadaso a albśmina en viales de uso śnico para ser reconstituido.
|
|
Efectos Colaterales:
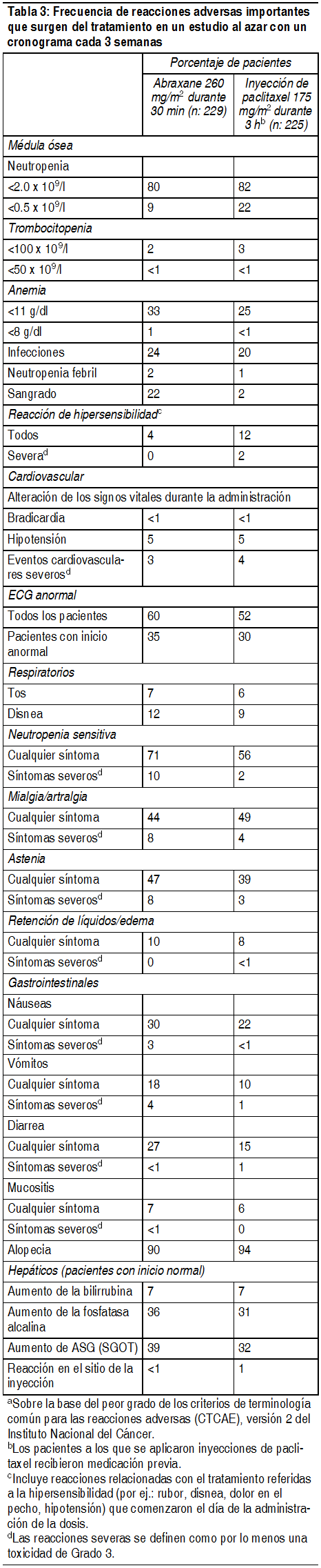
Dado que los ensayos clķnicos se llevan a cabo en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clķnicos de una droga no pueden compararse directamente con las tasas de los ensayos clķnicos de otra droga y pueden no reflejar las tasas observadas en la prįctica. Las reacciones adversas mįs comunes ( ³ 20%) con el uso śnico de Abraxane® en cįncer de mama metastįsico son alopecia, neutropenia, neuropatķa sensitiva, ECG anormal, fatiga/astenia, mialgia/artralgia, aumento de AST, aumento de la fosfatasa alcalina, anemia, nįusea, infecciones y diarrea. Las reacciones adversas mįs comunes ( ³ 20%) de Abraxane® en combinación con carboplatino para cįncer de pulmón no microcķtico son anemia, neutropenia, trombocitopenia, alopecia, neuropatķa periférica, nįuseas y fatiga. Las reacciones adversas severas mįs comunes de Abraxane® en combinación con carboplatino para cįncer de pulmón no microcķtico son anemia (4%) y neumonķa (3%). Las reacciones adversas mįs comunes que conllevaron a la discontinuación permanente de Abraxane® fueron neutropenia (3%), trombocitopenia (3%) y neuropatķa periférica (1%). Las reacciones adversas mįs comunes que conllevaron a la reducción de dosis de Abraxane® fueron neutropenia (24%), trombocitopenia (13%) y anemia (6%). Las reacciones adversas que conllevaron a la suspensión o retraso de las dosis de Abraxane® fueron neutropenia (41%), trombocitopenia (30%) y anemia (16%). En un estudio abierto, randomizado de Abraxane® en combinación con gemcitabina para adenocarcinoma pancreįtico (ver Estudios clķnicos), las reacciones adversas mįs frecuentes ( ³ 20%) seleccionadas (con una incidencia mįs elevada ³ 5%) de Abraxane® son neutropenia, fatiga, neuropatķa periférica, nįuseas, alopecia, edema periférico, diarrea pirexia, vómitos, disminución del apetito, sarpullido y deshidratación. Las reacciones adversas serias mįs frecuentes de Abraxane® (con una incidencia mįs elevada ³ 1%) son pirexia (6%), deshidratación (5%), neumonķa (4%) y vómitos (4%). Las reacciones adversas mįs frecuentes que resultan en la discontinuación permanente de Abraxane® son neuropatķa periférica (8%), fatiga (4%) y trombocitopenia (2%). Las reacciones adversas mįs frecuentes que resultan en la reducción de la dosis de Abraxane® son neutropenia (10%) y C. Las reacciones adversas mįs frecuentes que conducen a la suspensión o retraso en la dosis de Abraxane® son neutropenia (16%), trombocitopenia (12%), fatiga (8%), neuropatķa periférica (15%), anemia (5%) y diarrea (5%). Experiencia en ensayos clķnicos en cįncer de mama metastįsico: La tabla 6 muestra la frecuencia de las reacciones adversas importantes en un ensayo comparativo al azar para los pacientes que recibieron Abraxane® como śnico agente o una inyección de paclitaxel para el tratamiento del cįncer de mama metastįsico.
Ver Tabla
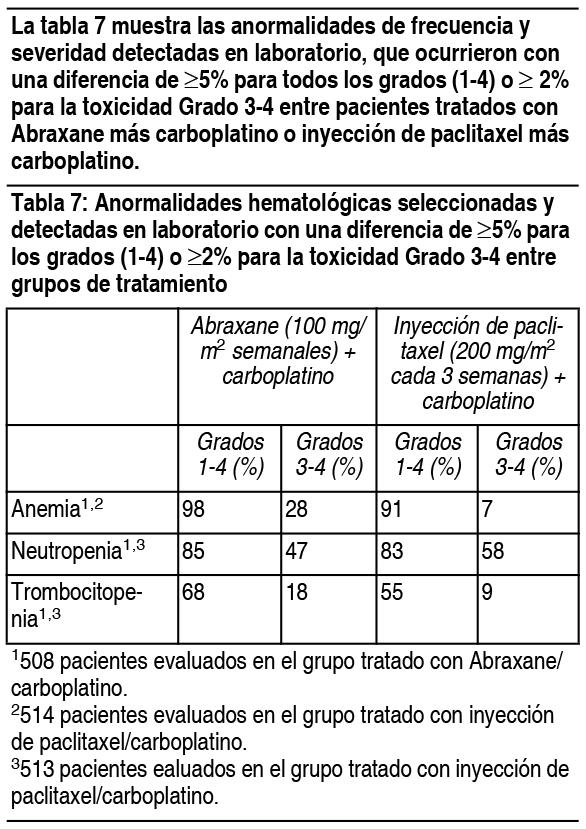
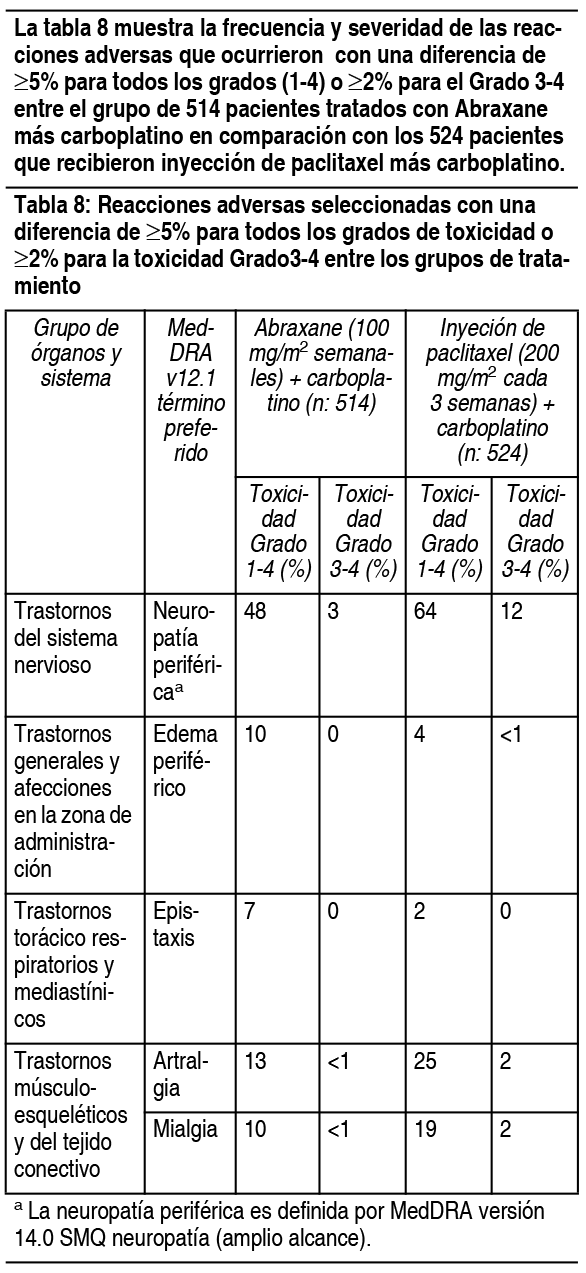
Experiencias de reacciones adversas por sistema corporal: Desórdenes hematológicos: La neutropenia dependió de la dosis y fue reversible. Entre los pacientes con cįncer de mama metastįsico de un ensayo al azar, el recuento de neutrófilos disminuyo a menos de 500 células/mm3 (Grado 4) en el 9% de los pacientes tratados con una dosis de 260 mg/m2, comparado con el 22% de los pacientes que recibieron una inyección de paclitaxel con una dosis de 175 mg/m2. Se ha observado pancitopenia en los ensayos clķnicos. Infecciones: Se informaron episodios infecciosos en el 24% de los pacientes tratados con Abraxane®. Las complicaciones infecciosas informadas con mįs frecuencia fueron candidiasis oral, infecciones de las vķas respiratorias y neumonķa. Reacciones de hipersensibilidad (HSRs): Se produjeron reacciones de hipersensibilidad de Grado 1 ó 2 el dķa de la administración de Abraxane®, que consistieron en disnea (1%) y rubor, hipotensión, dolor en el pecho y arritmia (todos <1%). No se estudió el uso de Abraxane® en pacientes que mostraron hipersensibilidad previa a la inyección de paclitaxel o a la albśmina humana. Cardiovasculares: Se produjo hipotensión, durante la infusión de 30 minutos, en el 5% de los pacientes. La bradicardia, durante la infusión de 30 minutos, se produjo en <1% de los pacientes. Estos cambios de los signos vitales la mayorķa de las veces no tuvieron sķntomas ni precisaron una terapia especķfica o la interrupción del tratamiento. Se produjeron reacciones cardiovasculares severas posiblemente relacionadas con el agente śnico Abraxane® en aproximadamente el 3% de los pacientes. Estas reacciones incluyeron isquemia/infarto cardķaco, dolor de pecho, paro cardķaco, taquicardia supra ventricular, edema, trombosis, tromboembolia pulmonar, embolia pulmonar e hipertensión. Se informaron casos de accidentes cerebrovasculares (apoplejķas) y ataques isquémicos transitorios. Las anormalidades de los electrocardiogramas (ECG) fueron comunes entre los pacientes al inicio. Las anormalidades de los ECG en estudio generalmente no produjeron sķntomas, no fueron limitantes de la dosis y no precisaron intervenciones. Se observaron anormalidades de ECG en el 60% de los pacientes. Entre los pacientes con ECG normal antes de ingresar al estudio, el 35% desarrollo una caracterķstica anormal mientras estuvo en el estudio. Las modificaciones de ECG informadas con mįs frecuencia fueron anormalidades de re polarización no especķficas, bradicardia sinusal y taquicardia sinusal. Respiratorios: Se informaron disnea (12%), tos (7%) y neumotórax (<1%) tras el tratamiento con Abraxane®. Neurológicos: La frecuencia y gravedad de la neuropatķa sensitiva aumentaron con la acumulación de la dosis. La neuropatķa sensitiva fue la causa de la interrupción de Abraxane® en 7/229 (3%) pacientes. Veinticuatro pacientes (10%) tratados con Abraxane® desarrollaron neuropatķa periférica de Grado 3; de estos pacientes, 14 tuvieron una mejora documentada tras un promedio de 22 dķas; 10 pacientes reanudaron el tratamiento con una dosis reducida de Abraxane® y 2 lo interrumpieron debido a una neuropatķa periférica. De los 10 pacientes sin mejorķa documentada, 4 interrumpieron el estudio debido a una neuropatķa periférica. No se observaron neuropatķas sensitivas de Grado 4. Solo se observó un incidente de neuropatķa motora (Grado 2) en uno de los brazos del ensayo controlado. Trastornos de la visión: Se produjeron disturbios oculares/visuales en el 13% de todos los pacientes (n=366) tratados con Abraxane® y el 1% fue severo. Se informaron casos severos (queratitis y visión borrosa) en pacientes que recibieron dosis mayores a las recomendadas (300 ó 375 mg/m2). Estos efectos generalmente fueron reversibles. Artralgia/mialgia: Los sķntomas generalmente fueron transitorios, se produjeron 2 ó 3 dķas después de la administración de Abraxane® y desaparecieron a los pocos dķas. Hepįticos: Se informaron aumentos de la GGT de Grado 3 ó 4 en el 14% de los pacientes tratados con Abraxane® y en el 10% de los pacientes tratados con una inyección de paclitaxel en un ensayo al azar. Renales: En total, el 11% de los pacientes tuvo un aumento de la creatinina; el 1% fue severo. Las toxicidades renales no produjeron interrupciones, reducciones o demoras de la dosis. Otras reacciones clķnicas: Se informaron cambios en las uńas (cambios en la pigmentación o decoloración de la base de las uńas). Se produjo edema en el 10% de los pacientes; ningśn paciente tuvo edema severo. También se informaron deshidratación y pirexia. Experiencia de ensayos clķnicos en cįncer de pulmón no microcķtico: Las reacciones adversas fueron evaluadas en 514 pacientes tratados con Abraxane®/carboplatino y en 524 pacientes tratados con inyección de paclitaxel/carboplatino que recibieron tratamiento sistémico de primera lķnea para cįncer de pulmón no microcķtico (NSCLC) localmente avanzado (etapa IIIB) o metastįsico (IV) en un ensayo abierto, multicéntrico y aleatorizado. Abraxane® fue administrado como infusión intravenosa durante 30 minutos con una dosis de 100 mg/m2 en los dķas 1, 8 y 15 de cada ciclo de 21 dķas. La inyección de paclitaxel fue administrada como infusión intravenosa durante 3 horas con una dosis de 200 mg/m2, luego de la premedicación. En ambos grupos de tratamiento, se administro carboplatino de forma intravenosa a una dosis de AUC = 6 mg · min/ml en el dķa 1 de cada ciclo de 21 dķas luego de finalizar la infusión de Abraxane®/paclitaxel. Las diferencias en la dosis y el esquema de paclitaxel entre los 2 grupos limitan la comparación directa de reacciones adversas dependientes de la dosis y el esquema. Entre los pacientes evaluables con respecto a las reacciones adversas, la mediana de edad era de 60 ańos, el 75% eran hombres y el 81% eran de raza blanca, el 49% tenķa adenocarcinoma, el 43% tenķa cįncer de pulmón de células escamosas, el 76% tenķa la escala ECOG PS 1. Los pacientes en ambos grupos de tratamiento recibieron una mediana de 6 ciclos de tratamiento. Las siguientes reacciones adversas comunes (incidencia ³ 10) fueron observadas a una incidencia similar en pacientes tratados con Abraxane® mįs carboplatino y tratados con inyección de paclitaxel mįs carboplatino: alopecia 56%, nįuseas 27%, fatiga 25%, apetito disminuido 17%, astenia 16%, constipación 16%, diarrea 15%, vómitos 12%, disnea 12% y rash 10% (las tasas de incidencia son para el grupo de tratamiento de Abraxane® mįs carboplatino).
Ver Tabla
Ver Tabla
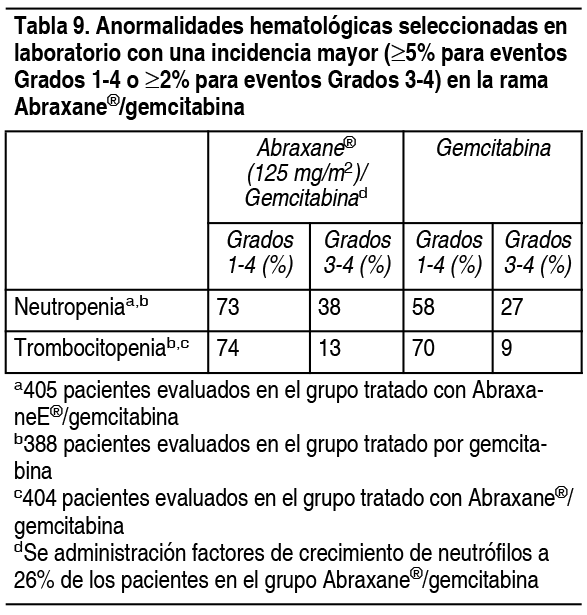
En el grupo tratado con Abraxane® mįs carboplatino, 17/514 (3%) pacientes desarrollaron neuropatķa periférica Grado 3 y ningśn paciente desarrollo neuropatķa periférica Grado 4. La neuropatķa Grado 3 mejoró a Grado 1 o se resolvió en 10/17 pacientes (59%) luego de la interrupción o discontinuación de Abraxane®. Experiencia en estudios clķnicos en adenocarcinoma del pįncreas: Se evaluaron reacciones adversas en 421 pacientes que recibieron Abraxane® mįs gemcitabina y 402 pacientes que recibieron gemcitabina para el tratamiento sistémico de primera lķnea adenocarcinoma metastįsico del pįncreas en un estudio abierto, controlado, randomizado, multinacional y multicéntrico. Los pacientes recibieron una mediana de duración del tratamiento de 3.9 meses en el grupo de Abraxane®/gemcitabina y 2.8 meses en el grupo de gemcitabina. Para la población tratada, la mediana de la intensidad de dosis relativa para la gemcitabina fue del 75% en el grupo de Abraxane®/gemcitabina y del 85% en el grupo de gemcitabina. La mediana de intensidad de dosis relativa de Abraxane® fue del 81%. La Tabla 9 muestra la frecuencia y la severidad de las anormalidades detectadas en laboratorio que ocurrieron a una incidencia mayor para toxicidades Grados 1-4 ( ³ 5%) o para toxicidad Grado 3-4 ( ³ 2%) en pacientes tratados con Abraxane® mįs gemcitabina.
Ver Tabla
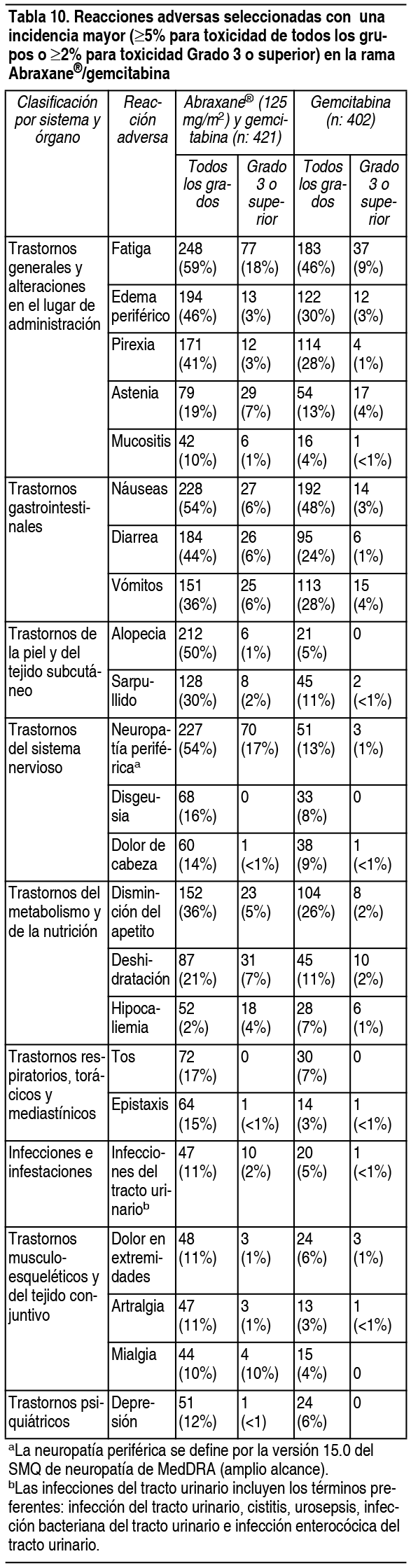
La Tabla 10 muestra la frecuencia y la severidad de reacciones adversas que ocurrieron con una diferencia ³ 5% para todos los grados o ³ 2% para Grado 3 o superior en el grupo tratado con Abraxane® mįs gemcitabina en comparación con el grupo con gemcitabina.
Ver Tabla
Las reacciones adversas clķnicamente relevantes adicionales que se informaron en <10% de los pacientes con adenocarcinoma del pįncreas que recibieron Abraxane®/gemcitabina incluyeron: Infecciones e infestaciones: candidiasis oral, neumonķa. Trastornos vasculares: hipertensión. Trastornos cardķacos: taquicardia, insuficiencia cardķaca congestiva. Trastornos oculares: edema macular cistoideo. Neuropatķa periférica: Se produjo neuropatķa periférica Grado 3 en 17% de los pacientes que recibieron Abraxane®/gemcitabina en comparación con el 1% de los pacientes que recibieron solo gemcitabina; ningśn paciente manifestó neuropatķa periférica Grado 4. La mediana del tiempo hasta la primera ocurrencia de neuropatķa periférica Grado 3 en la rama de Abraxane® fue de 140 dķas. Tras la suspensión de la dosis de Abraxane®, la mediana del tiempo hasta la mejorķa de neuropatķa periférica Grado 3 a £ Grado 1 fue de 29 dķas. De los pacientes tratados con Abraxane® neuropatķa periférica Grado 3, el 44% reanudaron Abraxane® a una dosis reducida. Sepsis: Se produjo sepsis en 5% de los pacientes que recibieron Abraxane/gemcitabina en comparación con 2% de los pacientes que recibieron solo gemcitabina. La sepsis se produjo tanto en pacientes con y sin neutropenia. Los factores de riesgo de sepsis incluyeron obstrucción biliar o presencia de un stent biliar. Neumonitis: Se produjo neumonitis en 4% de los pacientes que recibieron Abraxane®/gemcitabina en comparación con el 1% de los pacientes que recibieron gemcitabina sola. Dos de 17 pacientes en la rama de Abraxane® con neumonitis murieron. Experiencia tras la comercialización con Abraxane® y otras fórmulas de paclitaxel: A menos que se indique lo contrario, las referencias a continuación indican las reacciones adversas identificadas durante el uso tras la aprobación de Abraxane®. Dado que estas reacciones se informaron en forma voluntaria de una población de tamańo incierto, no siempre es posible calcular en forma confiable la frecuencia o establecer una relación causal a la exposición de la droga. En algunos casos, puede esperarse que con Abraxane® se produzcan reacciones severas observadas con la inyección de paclitaxel. Reacciones de hipersensibilidad: Se informaron reacciones de hipersensibilidad severas y algunas veces fatales con Abraxane®. No se ha estudiado el uso de Abraxane® en pacientes que previamente mostraron hipersensibilidad a la inyección de paclitaxel o a la albśmina humana. Cardiovasculares: Hubo informes de insuficiencia cardķaca congestiva y disfunción ventricular izquierda con Abraxane®. La mayorķa de las personas habķa recibido tratamiento previo con fįrmacos cardiotóxicos, como por ejemplo antraciclinas, o padecķa una enfermedad cardķaca subyacente. Respiratorias: Hubo informes de neumonitis, neumonķa intersticial y embolia pulmonar en pacientes que recibieron Abraxane®, informes de neumonitis por radiación en pacientes que recibieron radioterapia concurrente. Se recibieron informes de fibrosis pulmonar como parte de la supervisión continua de la seguridad de la inyección de paclitaxel y esta también puede observarse con Abraxane®. Neurológicas: Se informaron parįlisis nerviosa craneana y paresia de las cuerdas vocales, asķ como la produjo ķleo paralķtico. Trastornos de la visión: Los informes en la literatura de potenciales evocados visuales anormales en pacientes tratados con inyección de paclitaxel sugieren un dańo persistente del nervio óptico. Esto también puede observarse con Abraxane®. Ha sido reportada la reducción de la agudeza visual debido a edema macular cistoide (CME) durante el tratamiento con Abraxane® asķ como con otros taxanes. El CME mejora y la agudeza visual puede restituirse a su estado original luego de la suspensión del tratamiento. Hepįticas: Se recibieron informes de necrosis hepįtica y encefalopatķa hepįtica que produjeron la muerte como parte de la supervisión continua de la seguridad de la inyección de paclitaxel y esto puede ocurrir tras el tratamiento con Abraxane®. Gastrointestinales (GI): Hubo informes de obstrucción gastrointestinal, perforación intestinal, pancreatitis y colitis isquémica tras el tratamiento con Abraxane®. Hubo informes de enterocolitis neutropénica (tiflitis), a pesar de la administración concomitante de G-CSF, en los pacientes tratados solo con inyección de paclitaxel y en combinación con otros agentes quimioterįpicos. Reacción en el sitio de inyección: Hubo informes de extravasación de Abraxane®. Debido a la posibilidad de extravasación, se recomienda monitorear de cerca el lugar de infusión de Abraxane® por si esta se produce durante la administración del medicamento. Se informaron reacciones severas tales como flebitis, celulitis, endurecimiento, necrosis y fibrosis como parte de la supervisión continua de la seguridad de la inyección de paclitaxel. En algunos casos, el inicio de la reacción en el lugar de la inyección de paclitaxel en los pacientes inyectados se produjo durante una infusión prolongada o demoró de 1 semana a 10 dķas. Se informó recurrencia de reacciones cutįneas en el lugar de una extravasación previa tras la administración de la inyección de paclitaxel en un lugar diferente, es decir, "recuerdo". Otras reacciones clķnicas: Se observaron reacciones cutįneas, incluso erupción generalizada o maculopapular, eritema y prurito con Abraxane®. Hubo informes de casos de reacciones de fotosensibilidad, fenómeno de recuerdo de radiación y en algunos pacientes previamente expuestos a la capecitabina, se han notificado casos de eritrodisestesia palmar-plantar. Se informaron sķndrome de Stevens-Johnson y necrolisis epidérmica tóxica. Hubo informes de conjuntivitis, celulitis y aumento del lagrimeo con la inyección de paclitaxel. Exposición accidental: No se recibieron informes de exposición accidental a Abraxane®. Sin embargo, con la inhalación de paclitaxel se informaron disnea, dolor de pecho, ardor ocular, dolor de garganta y nįusea. Tras la exposición tópica, las reacciones incluyeron picazón, ardor y enrojecimiento.
|
|
Contraindicaciones:
Abraxane® no deberķa administrarse a pacientes con un recuento de neutrófilos inicial inferior a 1.500 células/mm3. La droga no deberķa administrarse nuevamente a pacientes que tuvieron una reacción de hipersensibilidad severa a Abraxane®.
|
|
Advertencias:
Efectos hematológicos: La supresión de la medula ósea (principalmente neutropenia) depende de la dosis y es una toxicidad limitante de dosis de Abraxane®. En estudios clķnicos, la neutropenia Grado 3-4 ocurrió en el 34% de pacientes con cįncer de mama metastįsico y en el 47% de pacientes con cįncer de pulmón no microcķtico y en el 38% de los pacientes con cįncer pancreįtico. Se debe realizar un monitoreo en busca de mielotoxicidad por medio de hemogramas completos de forma frecuente, incluido uno previo a la dosis en el dķa 1 (en caso de MBC) y en los dķas 1, 8 y 15 (en caso de NSCLC y cįncer pancreįtico). No administrar Abraxane® a pacientes con un recuento absoluto de neutrófilos (ANC) basal inferior a 1.500 células/mm3. En caso de presentarse neutropenia severa (<500 células/mm3 durante 7 dķas o mįs) en el transcurso del tratamiento con Abraxane®, reducir la dosis de Abraxane® en los ciclos posteriores en pacientes con MBC o NSCLC. En pacientes con MBC, reiniciar el tratamiento con ciclos cada 3 semanas de Abraxane® luego de recuperar el ANC a un nivel >1.500 células/mm3 y las plaquetas aun nivel >100.000 células/mm3. En pacientes con NSCLC, reiniciar el tratamiento si se lo recomienda (ver Posologķa, Tabla 2) con dosis reducidas permanentemente tanto para Abraxane® semanal como para carboplatino cada 3 semanas luego de recuperar el ANC a al menos 1500 células/mm3 y el recuento plaquetario a al menos 100.000 células/mm3 en el dķa 1 o el ANC a al menos 500 células/mm3 y el recuento plaquetario a al menos 50.000 células/mm3 en los dķas 8 o 15 del ciclo (ver Posologķa). En pacientes con adenocarcinoma del pįncreas, suspender Abraxane y gemcitabina si el ANC es menor de 500 células/mm3 o las plaquetas son menos de 50.000 células/mm3 y posponer el inicio del siguiente ciclo si el ANC es menor de 1500 células/mm3 o el recuento de plaquetas es menor de 100.000 células/mm3 en el dķa 1 del ciclo. Reanudar el tratamiento con la reducción de dosis apropiada si se recomienda (ver Posologķa). Sistema nervioso: La neuropatķa sensitiva es dependiente de la dosis y del esquema de dosis (ver Efectos colaterales). En general, ante la aparición de neuropatķa sensitiva de Grado 1 ó 2 no es necesario modificar la dosis. En caso de presentarse neuropatķa sensitiva ³ de Grado 3, se recomienda interrumpir el tratamiento hasta la resolución a Grado 1 ó 2 de cįncer de mama metastįsico o hasta la resolución a £ Grado 1 de NSCLC y cįncer pancreįtico, seguido de una reducción de la dosis para todos los ciclos posteriores de Abraxane® (ver Posologķa). Sepsis: Se observó sepsis en 5% de los pacientes con o sin neutropenia que recibieron Abraxane® en combinación con gemcitabina. La obstrucción biliar o la presencia de un stent biliar constituyeron factores de riesgo de sepsis severa o fatal. Si un paciente se vuelve febril (independientemente del ANC), iniciar el tratamiento con antibióticos de amplio espectro. Para la neutropenia febril, interrumpir Abraxane® y gemcitabina hasta que se resuelva la fiebre y el ANC sea ³ 1500; luego reanudar el tratamiento a niveles de dosis reducidos (ver Posologķa). Neumonitis: Se observó neumonitis, incluidos algunos casos que fueron fatales, en 4% de los pacientes que recibieron Abraxane® en combinación con gemcitabina. Monitorear a los pacientes para detectar signos y sķntomas de neumonitis e interrumpir Abraxane® y gemcitabina durante la evaluación de una presunta neumonitis. Luego de descartar una etiologķa infecciosa y tras realizar un diagnóstico de neumonitis, discontinuar permanentemente el tratamiento con Abraxane® y gemcitabina. Hipersensibilidad: Se han reportado reacciones de hipersensibilidad severas y en ocasiones fatales, incluyendo reacciones anafilįcticas. En los pacientes que hayan experimentado reacciones severas de hipersensibilidad a Abraxane® no debe reiniciarse el tratamiento con esta droga. Insuficiencia hepįtica: Dado que la exposición y la toxicidad de paclitaxel pueden aumentar con la insuficiencia hepįtica, la administración de Abraxane® en pacientes con esta insuficiencia debe realizarse con precaución. Los pacientes con insuficiencia hepįtica pueden tener mayor riesgo de toxicidad, particularmente mielosupresión; estos pacientes deben monitorearse minuciosamente para detectar el desarrollo de mielosupresión profunda. No se recomienda Abraxane en pacientes que tengan una bilirrubina total >5 x ULN. Ademįs, no se recomienda Abraxane en pacientes con adenocarcinoma de pįncreas metastįsico que tengan insuficiencia hepįtica moderada a severa (bilirrubina total >1.5 x ULN y AST £ 10 x ULN). La dosis inicial debe reducirse en pacientes que padezcan insuficiencia hepįtica moderada y severa (ver Posologķa, Uso en poblaciones especķficas y Farmacologķa clķnica). Albśmina (humana): Abraxane® contiene albśmina (humana), un derivado de la sangre humana. Sobre la base de una selección eficiente de donantes y de procesos de elaboración del producto, existe un riesgo remoto de transmisión de enfermedades virales. También se considera extremamente remoto un riesgo teórico de transmisión de la enfermedad de Creutzfeldt-Jakob (CJD). No se han identificado casos de transmisión de enfermedades virales o CJD para la albumina. Uso en el embarazo: Abraxane® puede causar danos al feto cuando se administra a una mujer embarazada. La administración de partķculas de paclitaxel unidas a proteķnas a ratas durante la preńez en dosis inferiores a la dosis mįxima recomendada en humanos, sobre la base de la superficie corporal, provocó toxicidad fetal, incluso mortalidad intrauterina, aumento de reabsorciones, reducción del nśmero de fetos vivos y malformaciones. No existen estudios adecuados y bien controlados en mujeres embarazadas que reciben Abraxane®. Si este fįrmaco se usa durante el embarazo o si la paciente queda embarazada mientras lo recibe, debe informarse a la paciente sobre el posible peligro para el feto. Debe advertirse a las mujeres en edad fértil que eviten quedar embarazadas mientras reciben Abraxane® (ver Uso en poblaciones especķficas). Uso en hombres: Debe advertirse a los hombres que eviten engendrar un hijo mientras reciben Abraxane® (ver Toxicologķa no clķnica).
|
|
Precauciones:
Uso en poblaciones especķficas: Embarazo: Embarazo categorķa D (ver Advertencias). No existen estudios adecuados y bien controlados en mujeres embarazadas que usan Abraxane®. Sobre la base de su mecanismo de acción y los descubrimientos en animales, Abraxane® puede causar dańo fetal al administrarse a una mujer embarazada. Si este medicamento se usa durante el embarazo o si la paciente se embaraza mientras recibe esta droga, debe advertķrsele sobre el posible peligro para el feto. Debe advertirse a las mujeres en edad fértil que eviten quedar embarazadas mientras reciben Abraxane®. La administración a ratas de partķculas de paclitaxel unidas a proteķnas durante la preńez, en los dķas de gestación 7 a 17 con dosis de 6 mg/m2 (aproximadamente el 2% de la dosis diaria mįxima recomendada en humanos sobre la base de mg/m2) provocaron toxicidad fetal, segśn indica la mortalidad intrauterina, aumento de las reabsorciones (hasta 5 veces), reducción del nśmero de fetos vivos y malformaciones, reducción del peso corporal fetal y aumento de las malformaciones fetales. Las anormalidades fetales incluyeron malformaciones de los tejidos blandos y esqueléticos, tales como ojo abultado, retina doblada, microftalmia y dilatación de los ventrķculos cerebrales. También se produjo una incidencia menor de malformaciones de los tejidos blandos y esqueléticos con 3 mg/m2 (aproximadamente el 1% de la dosis diaria mįxima recomendada en humanos sobre la base de mg/m2). Lactancia: Se desconoce si paclitaxel se excreta en la leche materna. Paclitaxel y/o sus metabolitos se excretaron en la leche de ratas lactantes. Dado que muchas drogas se excretan en la leche humana y debido a la posibilidad de reacciones adversas severas en lactantes, debe tomarse una decisión respecto a la continuidad o la interrupción del fįrmaco, teniendo en cuenta la importancia de la droga para la madre. Uso pediįtrico: No se han evaluado la seguridad y la eficacia de Abraxane® en pacientes pediįtricos. Uso geriįtrico: De los 229 pacientes de un estudio al azar que recibieron Abraxane® para el tratamiento de cįncer de mama metastįsico, el 13% tenķa por lo menos 65 ańos de edad y < 2% tenķa 75 ańos o mįs. No se produjeron toxicidades notablemente mįs frecuentes entre los pacientes que recibieron Abraxane®. Posteriormente se realizó un anįlisis agrupado con 981 pacientes que recibieron Abraxane® como monoterapia para cįncer de mama metastįsico, de los cuales el 15% tenķan 65 ańos o mįs y el 2% tenķan 75 ańos o mįs. En los pacientes de 65 ańos o mayores se observó una mayor incidencia de epistaxis, diarrea, deshidratación, fatiga y edema periférico. De los 514 pacientes en el estudio aleatorizado que recibieron Abraxane® y carboplatino para el tratamiento de primera lķnea de cįncer de pulmón no microcķtico, el 31% tenķa 65 ańos de edad o mįs y el 3.5% tenķan 75 ańos de edad o mįs. La mielosupresión, neuropatķa periférica y artralgia fueron mįs frecuentes en pacientes de 65 ańos o mįs en comparación con pacientes de menos de 65 ańos. No se observó una diferencia total en efectividad, tal como se midió por medio de tasas de respuesta entre pacientes de 65 ańos o mįs en comparación con pacientes de menos de 65 ańos. De los 431 pacientes en el estudio randomizado que recibieron Abraxane® y gemcitabina para el tratamiento de primera lķnea de adenocarcinoma pancreįtico, el 41% tenķa 65 ańos de edad o mįs y el 10% tenķa 75 ańos o mįs. No se observaron diferencias totales en la efectividad entre pacientes que tenķan 65 ańos o mįs y pacientes mįs jóvenes. En los pacientes de 65 ańos o mįs fue mįs frecuente diarrea, disminución del apetito, deshidratación y epistaxis que en los pacientes menores de 65 ańos. Los estudios clķnicos de Abraxane® no incluyeron una cantidad suficiente de pacientes con cįncer pancreįtico que tuviera 75 ańos y mįs para determinar si responden de manera diferente que los pacientes mįs jóvenes. Pacientes con insuficiencia hepįtica: La exposición al paclitaxel puede ser mayor en pacientes con insuficiencia hepįtica que en los pacientes con función hepįtica normal. La dosis inicial de Abraxane® debe reducirse en pacientes con insuficiencia hepįtica moderada a severa. No administrar Abraxane® en pacientes con una bilirrubina total >5 x ULN o una AST >10 x ULN (ver Posologķa, Advertencias y Farmacologķa clķnica). No administrar en pacientes con adenocarcinoma metastįsico del pįncreas que tengan insuficiencia hepįtica moderada a severa (ver Posologķa). Pacientes con insuficiencia renal: No se ha estudiado el uso de Abraxane® en pacientes con insuficiencia renal leve a moderada (aclaramiento de creatinina calculado ³ 30 a <90 ml/min) (ver Farmacologķa clķnica). Los datos son insuficientes como para permitir recomendaciones de dosificación en pacientes con insuficiencia renal severa o enfermedad renal terminal (aclaramiento de creatinina calculado <30 ml/min).
|
|
Interacciones Medicamentosas:
El metabolismo de paclitaxel es catalizado por las isoenzimas CYP2C8 y CYP3A4. En ausencia de estudios clķnicos formales de interacciones medicamentosas, debe tenerse cuidado al administrar Abraxane® conjuntamente con medicamentos inhibidores (por ejemplo: ketoconazol y otros antifśngicos imidazólicos, eritromicina, fluoxetina, gemfibrozilo, cimetidina, ritonavir, saquinavir, indinavir y nelfinavir) o inductores (por ejemplo: rifampicina, carbamazepina, fenitoķna, efavirenz, nevirapina) conocidos de CYP2C8 o de CYP3A4.
|
|
Sobredosificación:
No se conocen antķdotos para la sobredosis de Abraxane®. Las principales complicaciones previstas de la sobredosis serian la mielosupresión de la médula ósea, neuropatķa periférica y mucositis. Ante la eventualidad de una sobredosificación concurrir al hospital mįs cercano o comunicarse con Tecnofarma al télefono 225949201 o a la dirección electrónica: www.tecnofarma.cl
|
|
Conservación:
Almacenar a temperatura no mayor de 30° C. Conservar en el envase original para protegerlo de la luz intensa.
|
|
Observaciones:
Mantener fuera del alcance de los nińos.
|
|
Presentaciones:
Envase conteniendo 1 frasco-ampolla.
|
|



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}