 |
Composiciµn:
Cada jeringa prellenada contiene: Pegfilgrastim ADNr 6.00 mg, Acido AcÕtico Glacial, Sorbitol, Polisorbato 20, Hidrµxido de Sodio.
|
|
Acciµn TerapÕutica:
Inmunoestimulantes, factores estimulantes de colonias.
|
|
Indicaciones:
Reducciµn de la duraciµn de la neutropenia y de la incidencia de neutropenia febril en pacientes adultos con tumores malignos tratados con quimioterapia citotµxica (con excepciµn de leucemia mieloide crµnica y sÚndromes mieloplÃsicos).
|
|
Propiedades:
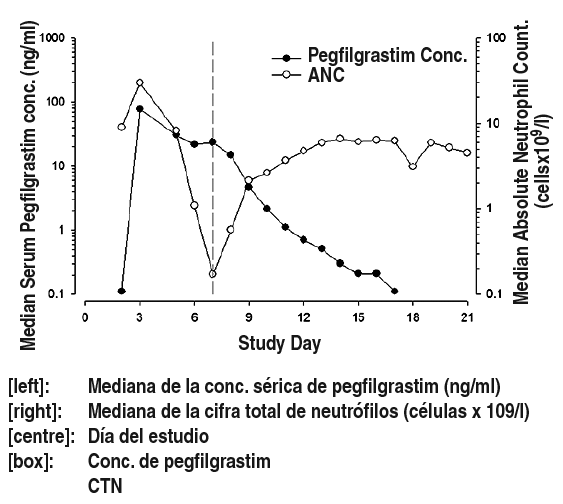
Acciµn (farmacologÚa y/o terapÕutica) a los modos de acciµn del medicamento en el hombre: FarmacologÚa: El factor humano estimulante de colonias de granulocitos (FEC-G) es una glucoproteÚna que regula la producciµn y liberaciµn de neutrµfilos desde la mÕdula µsea. Pegfilgrastim es un conjugado covalente del FEC-G humano recombinante (r-metHuG-CSF) con una molÕcula de polietilenglicol (PEG) de 20 kd. Pegfilgrastim es una forma de duraciµn sostenida de filgrastim como consecuencia de una menor eliminaciµn renal. Pegfilgrastim y filgrastim presentan el mismo mecanismo de acciµn, causando un aumento marcado de los neutrµfilos en sangre perifÕrica en 24 horas, con elevaciones mÚnimas de los monocitos y/o linfocitos. Similar a filgrastim, los neutrµfilos producidos en respuesta a pegfilgrastim presentan una funcionalidad normal o mejorada como demuestran las pruebas de quimiotaxis y de funciµn fagocÚtica. Al igual que otros factores de crecimiento hematopoyÕticos, el FEC-G ha demostrado propiedades estimuladoras sobre las cÕlulas endoteliales humanas in vitro. El FEC-G puede promover el crecimiento de las cÕlulas mieloides, incluyendo las cÕlulas tumorales y pueden observarse efectos similares en algunas cÕlulas no mieloides in vitro. En 2 estudios pivotales, doble ciego con asignaciµn aleatoria, en pacientes con cÃncer de mama de alto riesgo, etapa II-IV, tratados con quimioterapia mielosupresora consistente en doxorubicina y docetaxel, el uso de pegfilgrastim, como dosis ºnica 1 vez por ciclo, redujo la duraciµn de la neutropenia y la incidencia de neutropenia febril de forma similar a la observada con la administraciµn diaria de filgrastim (una mediana de 11 dÚas de administraciµn). En ausencia de soporte con factor de crecimiento, se ha descrito que este rÕgimen de quimioterapia suele resultar en una duraciµn media de la neutropenia de grado 4 de 5 a 7 dÚas, y de un 30-40% de incidencia de neutropenia febril. En un ensayo (n = 157) que usµ una dosis fija de 6 mg de pegfilgrastim, la duraciµn media de la neutropenia grado 4 para el grupo tratado con pegfilgrastim fue de 1.8 dÚas comparado con 1.6 dÚas del grupo tratado con filgrastim (0.23 dÚas de diferencia, IC de 95%: -0.15; 0.63). Durante el ensayo completo, el porcentaje de neutropenia febril fue de 13% en los pacientes tratados con pegfilgrastim comparado con 20% de los pacientes tratados con filgrastim (diferencia del 7%, IC de 95%: -19%; 5%). En el segundo ensayo (n = 310) en el que se usµ una dosis ajustada segºn el peso (100 mcg/kg), la duraciµn media de la neutropenia grado 4 en el grupo tratado con pegfilgrastim fue de 1.7 dÚas, comparado con 1.8 dÚas en el grupo tratado con filgrastim (diferencia de 0.03 dÚas, IC de 95%: -0.36; 0.30). El porcentaje total de neutropenia febril fue del 9% en los pacientes tratados con pegfilgrastim y de 18% de los pacientes tratados con filgrastim (diferencia 9%, IC de 95%: -16.8%; -1.1%). Se evaluµ en un ensayo clÚnico doble ciego, controlado con placebo, en pacientes con cÃncer de mama, el efecto de pegfilgrastim sobre la incidencia de neutropenia febril, tras la administraciµn de un rÕgimen de quimioterapia asociado a una tasa de neutropenia febril de 10-20% (docetaxel 100 mg/m2 cada 3 semanas durante 4 ciclos). Se asignaron aleatoriamente 928 pacientes para recibir una dosis ºnica de pegfilgrastim o de placebo, aproximadamente 24 horas (DÚa 2) posterior a la administraciµn de quimioterapia en cada ciclo. La incidencia de neutropenia febril fue menor en los pacientes que recibieron pegfilgrastim comparado con placebo (1% versus 17%, p< 0.001). La incidencia de hospitalizaciones y uso de antibiµticos IV asociados con un diagnµstico clÚnico de neutropenia febril, fue menor en el grupo de pegfilgrastim comparado con el del placebo (1% versus 14%, p< 0.001; y 2% versus 10%, p< 0.001). En un ensayo de fase II, doble ciego, de asignaciµn aleatoria, con un nºmero reducido de pacientes (n = 83) con leucemia mieloide aguda de novo que recibÚan quimioterapia, se comparµ pegfilgrastim (dosis ºnica de 6 mg) con filgrastim administrados durante la quimioterapia de inducciµn. La mediana del tiempo de recuperaciµn de la neutropenia grave fue de aproximadamente 22 dÚas en ambos grupos de tratamiento. No se estudiaron los efectos a largo plazo (ver Precauciones). En un ensayo fase II (n = 37), abierto, multicÕntrico, de asignaciµn aleatoria en pacientes pediÃtricos con sarcoma, que recibieron 100 çg/kg de pegfilgrastim tras un ciclo de quimioterapia con vincristina, doxorubicina y ciclofosfamida (VAdriaC/IE), se observµ una mayor duraciµn de la neutropenia grave (neutrµfilos <0.5 x 109) en niþos menores entre 0-5 aþos (8,9 dÚas), comparado con niþos de mayor edad, entre 6-11 aþos y entre 12-21 aþos (6 dÚas y 3.7 dÚas, respectivamente) y adultos. Adicionalmente, se observµ mayor incidencia de neutropenia febril en niþos menores entre 0-5 aþos (75%) comparado con niþos de mayor edad entre 6-11 aþos y entre 12-21 aþos (70% y 33%, respectivamente) y adultos (ver Efectos colaterales y FarmacocinÕtica). FarmacocinÕtica: Tras una sola dosis subcutÃnea de pegfilgrastim, la concentraciµn sÕrica mÃxima ocurre de 16 a 120 horas despuÕs de la administraciµn y las concentraciones sÕricas de pegfilgrastim se mantienen durante el perÚodo de neutropenia posterior a la quimioterapia mielosupresora. La eliminaciµn de pegfilgrastim es no lineal con respecto a la dosis; el aclaramiento o eliminaciµn sÕrica de pegfilgrastim disminuye al aumentar la dosis. Pegfilgrastim parece eliminarse principalmente por el aclaramiento mediado por neutrµfilos, que se satura a altas dosis. Consistente con un mecanismo de aclaramiento autorregulado, la concentraciµn sÕrica de pegfilgrastim disminuye rÃpidamente al comenzar la recuperaciµn de los neutrµfilos (ver figura 1). Figura 1: Perfil de la mediana en la concentraciµn sÕrica de pegfilgrastim y la cuenta Absoluta de Neutrµfilos (CAN) en pacientes tratados con quimioterapia despuÕs de la administraciµn de una ºnica inyecciµn de 6 mg.
Ver Tabla
Debido al mecanismo de aclaramiento regulado por neutrµfilos, no se espera que la farmacocinÕtica de pegfilgrastim se vea afectada por insuficiencia renal o hepÃtica. En un ensayo abierto de dosis ºnica (n = 31), las diferentes etapas de la insuficiencia renal, incluyendo insuficiencia renal con requerimientos de diÃlisis no tuvieron impacto sobre la farmacocinÕtica de pegfilgrastim. Personas de edad avanzada: Los escasos datos disponibles indican que la farmacocinÕtica de pegfilgrastim en personas de edad avanzada (> 65 aþos) es similar a la de los adultos. Poblaciµn pediÃtrica: La farmacocinÕtica de pegfilgrastim se estudiµ en 37 pacientes pediÃtricos con sarcoma, quienes recibieron 100 mcg/kg de pegfilgrastim tras completar la quimioterapia con VAdriaC/IE. El grupo de menor edad (0-5 aþos) presentµ una media de exposiciµn mÃs alta a pegfilgrastim (AUC) (Ý Desviaciµn EstÃndar) (47.9 Ý 22.5 mcg ñ hr/ml) que los niþos de mayor edad entre 6-11 aþos y entre 12-21 aþos (22.0 Ý 13.1 mcg ñ hr/ml y 29.3 Ý 23.2 mcg ñ hr/ml, respectivamente) (ver FarmacologÚa). Con la excepciµn del grupo de edad mÃs joven (0-5 aþos), la AUC media en pacientes pediÃtricos fue similar a la de pacientes adultos con cÃncer de mama de alto riesgo en etapas II-IV que recibieron 100 mcg/kg de pegfilgrastim despuÕs de finalizar el tratamiento con doxorubicina/docetaxel (ver Efectos colaterales y FarmacocinÕtica).
|
|
PosologÚa:
VÚa de administraciµn y dosificaciµn: El tratamiento con Neulastim debe ser iniciado y supervisado por un mÕdico con experiencia en oncologÚa y/o hematologÚa. PosologÚa: La dosis recomendada de Neulastim es de 6 mg (una jeringa prellenada) por cada ciclo de quimioterapia, administrada 24 horas despuÕs de la quimioterapia citotµxica. MÕtodo de administraciµn: Neulastim se administra por inyecciµn subcutÃnea. Las inyecciones se deben administrar en el muslo, el abdomen o en la parte superior del brazo. Poblaciµn pediÃtrica: No se han establecido aºn la seguridad y eficacia de Neulastim en niþos. En Efectos colaterales, FarmacologÚa y FarmacocinÕtica, se describen los datos disponibles actualmente, sin embargo, no se puede hacer una recomendaciµn posolµgica. Pacientes con insuficiencia renal: No se recomienda modificar la dosis en pacientes con insuficiencia renal, incluyendo aquellos con insuficiencia renal crµnica terminal.
|
|
Modo de Empleo:
Instrucciones especiales de uso, manipulaciµn y eliminaciµn: Las jeringas precargadas de Neulastim son para un solo uso (desechables). Neulastim es una soluciµn estÕril sin conservantes. Antes de administrar la soluciµn de Neulastim, se debe comprobar que no contiene partÚculas visibles. Sµlo se inyectarà si està lÚmpida y es incolora. Si se agita en exceso, el pegfilgrastim puede agregarse y perder la actividad biolµgica. Antes de inyectar el medicamento, se debe dejar que la jeringa precargada alcance la temperatura ambiente. El producto que no se haya utilizado y el material de desecho deben eliminarse con arreglo a la normativa local. Eliminaciµn de medicamentos no utilizados/caducados: La emisiµn de productos farmacÕuticos al medio ambiente debe reducirse a un mÚnimo. Los medicamentos no deben eliminarse a travÕs de las aguas residuales, y su eliminaciµn con los residuos domÕsticos tambiÕn debe evitarse. Utilice los sistemas de recogida establecidos si los hay en su localidad.
|
|
Efectos Colaterales:
Resumen del perfil de seguridad: Las reacciones adversas notificadas con mayor frecuencia fueron dolor µseo (muy frecuentes [ ° 1/10]) y dolor mºsculo-esquelÕtico (muy frecuentes). El dolor µseo fue generalmente de intensidad leve a moderada, transitorio y en la mayorÚa de los pacientes se controlµ con analgÕsicos comunes. En tratamientos iniciales o subsecuentes con Neulastim, se han observado reacciones de hipersensibilidad, incluyendo erupciones cutÃneas, urticaria, angioedema, disnea, eritema, rubor e hipotensiµn (poco frecuentes [ ° 1/1.000 a < 1/100]). En pacientes en tratamiento con Neulastim pueden ocurrir reacciones alÕrgicas graves, incluyendo anafilaxis (poco frecuentes) (ver Precauciones). SÚndrome de fuga capilar, el cual puede poner en peligro la vida si se retrasa el tratamiento; se ha notificado raramente ( ° 1/10.000 a < 1/1.000) en pacientes con cÃncer sometidos a quimioterapia tras la administraciµn de factores estimulantes de colonias de granulocitos; ver Precauciones y secciµn "Descripciµn de las reacciones adversas seleccionadas" a continuaciµn. Esplenomegalia, generalmente asintomÃtica, es poco frecuente. Se han notificado casos poco frecuentes de ruptura esplÕnica, incluyendo algunos casos fatales, tras la administraciµn de pegfilgrastim (ver Precauciones). Se han notificado reacciones adversas pulmonares poco frecuentes incluyendo neumonÚa intersticial, edema pulmonar, infiltrados pulmonares y fibrosis pulmonar. Casos poco frecuentes han resultado en insuficiencia respiratoria o en el sÚndrome de distrÕs respiratorio agudo (SDRA), potencialmente mortal (ver Precauciones). Se han notificado casos aislados de crisis de cÕlulas falciformes en pacientes con rasgo de cÕlulas falciformes o anemia de cÕlulas falciformes (ver Precauciones). Tabla de reacciones adversas: Los datos incluidos en la tabla siguiente describen las reacciones adversas notificadas durante ensayos clÚnicos y de notificaciones espontÃneas. Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia. Reacciones adversas: Muy frecuentes ( ° 1/10); Frecuentes ( ° 1/100 a < 1/10); Poco frecuentes ( ° 1/1.000 a < 1/100); Raras ( ° 1/10.000 a < 1/1.000); Muy raras (< 1/10.000). Trastornos de la sangre y del sistema linfÃtico: Frecuentes: Trombocitopenia1. Poco frecuentes: Crisis de cÕlulas falciformes2. Leucocitosis1. Esplenomegalia2. Ruptura esplÕnica2. Trastornos del sistema inmunolµgico: Poco frecuentes: Reacciones de hipersensibilidad; Anafilaxis. Trastornos del metabolismo y de la nutriciµn: Poco frecuentes: Elevaciµn del Ãcido ºrico. Trastornos del sistema nervioso: Muy frecuentes: Cefalea1. Trastornos vasculares: Raras: SÚndrome de fuga capilar1. Trastornos respiratorios, torÃcicos y mediastÚnicos: Poco frecuentes: SÚndrome de distrÕs respiratorio del adulto2. Reacciones adversas pulmonares (neumonÚa intersticial, edema pulmonar, infiltrados pulmonares y fibrosis pulmonar). Trastornos gastrointestinales: Muy frecuentes: NÃusea1. Trastornos de la piel y del tejido subcutÃneo: Poco frecuentes: SÚndrome de Sweet (dermatosis febril aguda)1,2; Vasculitis cutÃnea1,2. Trastornos mºsculo-esquelÕticos y del tejido conjuntivo: Muy frecuentes: Dolor µseo; Dolor mºsculo-esquelÕtico (mialgia, artralgia, dolor en las extremidades, dolor en la espalda, dolor mºsculo-esquelÕtico, dolor en el cuello). Trastornos generales y alteraciones en el lugar de administraciµn: Frecuentes: Reacciµn en el sitio de la inyecciµn (incluyendo dolor)1. Poco frecuentes: Dolor torÃcico no cardÚaco. Exploraciones complementarias: Poco frecuentes: Incremento en la lactato deshidrogenasa y fosfatasa alcalina1; Elevaciones transitorias en las pruebas de funciµn hepÃtica para ALT o AST1. 1Ver secciµn "Descripciµn de las reacciones adversas seleccionadas" mÃs adelante. 2 Se ha identificado este evento adverso en la monitorizaciµn postcomercializaciµn, pero no se ha observado la misma en los ensayos clÚnicos controlados aleatorizados en adultos que respaldaron la autorizaciµn de comercializaciµn. Se ha estimado la frecuencia de la categorÚa de acuerdo con un cÃlculo estadÚstico basado en 932 pacientes que recibieron Neulastim en 7 ensayos clÚnicos aleatorizados. Descripciµn de las reacciones adversas seleccionadas: Se han notificado casos poco frecuentes de sÚndrome de Sweet, aunque en algunos casos las enfermedades hematolµgicas malignas subyacentes pueden estar relacionadas con su apariciµn. Se han notificado acontecimientos poco frecuentes de vasculitis cutÃnea en pacientes tratados con Neulastim. Se desconoce el mecanismo de apariciµn de vasculitis en pacientes que reciben Neulastim. De forma inicial o en tratamientos posteriores a Neulastim, se han notificado (frecuentemente [ ° 1/100 a <1/10]) reacciones en el sitio de la inyecciµn, incluyendo dolor y eritema. Se han notificado casos poco frecuentes de leucocitosis (cuenta de leucocitos > 100 x 109/l) (ver Precauciones). Elevaciones reversibles, leves a moderadas en Ãcido ºrico y en fosfatasa alcalina, sin efectos clÚnicos asociados; y elevaciones reversibles en lacto deshidrogenasa, sin efectos clÚnicos asociados fueron poco frecuentes en pacientes tratados con Neulastim despuÕs de la quimioterapia citotµxica. Se observµ de manera muy frecuente nÃusea y cefalea en los pacientes tratados con quimioterapia. Elevaciones poco frecuentes en las pruebas de la funciµn hepÃtica: ALT (alanina aminotransferasa) o AST (aspartato aminotransferasa), han sido observadas en pacientes despuÕs de recibir tratamiento con pegfilgrastim posterior a la quimioterapia citotµxica. Estas elevaciones son transitorias y retornan al estado basal. Se han notificado casos frecuentes de trombocitopenia. Se han notificado casos de sÚndrome de fuga capilar durante la postcomercializaciµn con el uso de factores estimulantes de colonias de granulocitos. Estos casos ocurrieron generalmente en pacientes con enfermedades neoplÃsicas malignas avanzadas, sepsis, que reciben mºltiples quimioterapÕuticos o sometidos a afÕresis (ver Precauciones). Poblaciµn pediÃtrica: La experiencia en niþos es limitada. Se ha observado mayor frecuencia de reacciones adversas graves en niþos menores, entre 0-5 aþos (92%) comparado con niþos de mayor edad, entre 6-11 aþos y 12-21 aþos respectivamente (80% y 67%), y adultos. La reacciµn adversa notificada con mayor frecuencia fue dolor µseo (ver FarmacologÚa y FarmacocinÕtica).
|
|
Contraindicaciones:
Hipersensibilidad al principio activo o a alguno de los excipientes.
|
|
Advertencias:
Neulastim contiene sorbitol. Pacientes con problemas hereditarios de intolerancia a la fructosa, no deberÚan ser tratados con este medicamento. Neulastim contiene menos de 1 mmol (23 mg) de sodio por 6 mg de dosis, por lo que se considera "exento de sodio". Para mejorar la trazabilidad de los factores estimulantes de colonias de granulocitos (FEC-G), la marca comercial del producto administrado debe estar correctamente registrada en la historia clÚnica del paciente.
|
|
Precauciones:
Datos clÚnicos obtenidos a partir de un nºmero limitado de pacientes sugieren que pegfilgrastim tiene un efecto comparable a filgrastim en el tiempo de recuperaciµn de la neutropenia grave en pacientes con leucemia mieloide aguda de novo (ver FarmacologÚa). Sin embargo, no se han establecido los efectos a largo plazo de Neulastim en la leucemia mieloide aguda; por lo tanto, se debe usar con precauciµn en esta poblaciµn de pacientes. Los factores estimulantes de colonias de granulocitos pueden estimular el crecimiento de cÕlulas mieloides in vitro y podrÚan observarse efectos similares en algunas cÕlulas no mieloides in vitro. No se ha investigado la seguridad y eficacia de Neulastim en pacientes con sÚndrome mielodisplÃsico, leucemia mieloide crµnica, ni en pacientes con leucemia mieloide aguda (LMA) secundaria; por lo tanto no debe utilizarse en estos pacientes. Se debe tener especial precauciµn al distinguir el diagnµstico de la transformaciµn blÃstica de la leucemia mieloide crµnica de una leucemia mieloide aguda. No se han establecido la seguridad y la eficacia de Neulastim administrado en pacientes menores de 55 aþos con leucemia mieloide aguda de novo con citogenÕtica t(15;17). No se han investigado la seguridad y la eficacia de Neulastim en pacientes tratados con dosis altas de quimioterapia. Este medicamento no debe utilizarse para incrementar las dosis de la quimioterapia citotµxica por encima de los regÚmenes posolµgicos establecidos. Se han notificado reacciones adversas pulmonares poco frecuentes ( ° 1/1.000 a < 1/100), tras la administraciµn de factores estimulantes de colonias de granulocitos (FEC-G), en particular neumonÚa intersticial. Pacientes con antecedente reciente de infiltrados pulmonares o neumonÚa pueden estar en mayor riesgo (ver Efectos colaterales). La apariciµn de sÚntomas respiratorios tales como tos, fiebre y disnea, en asociaciµn con signos radiolµgicos de infiltraciµn y deterioro de la funciµn pulmonar, junto con un aumento en la cuenta de neutrµfilos pueden ser signos preliminares del sÚndrome de distrÕs respiratorio en el adulto (SDRA). En estos casos, se deberà suspender la administraciµn de Neulastim, a discreciµn del mÕdico, y administrar el tratamiento apropiado (ver Efectos colaterales). Se ha notificado sÚndrome de fuga capilar tras la administraciµn de factores estimulantes de colonias de granulocitos, que se caracteriza por hipotensiµn, hipoalbuminemia, edema y hemoconcentraciµn. Los pacientes que desarrollen sÚntomas del sÚndrome de fuga capilar deben monitorearse cuidadosamente y deben recibir tratamiento de soporte estÃndar, que puede incluir la necesidad de cuidados intensivos (ver Efectos colaterales). Se han notificado casos poco frecuentes y generalmente asintomÃticos de esplenomegalia y casos poco frecuentes de ruptura esplÕnica despuÕs de la administraciµn de pegfilgrastim, incluyendo algunos casos mortales (ver Efectos colaterales). Por lo tanto, el tamaþo del bazo debe evaluarse cuidadosamente (p.ej., examen clÚnico, ultrasonido). Se deberà considerar el diagnµstico de ruptura esplÕnica en pacientes que reporten dolor en la parte superior izquierda del abdomen o en el extremo del hombro. El tratamiento con Neulastim solo, no evita la trombocitopenia ni la anemia debidas al mantenimiento de dosis completas de quimioterapia mielosupresora en el esquema prescrito. Se recomienda evaluar regularmente la cuenta de plaquetas y el hematocrito. Se debe tener especial cuidado cuando se administren agentes quimioterapÕuticos en monoterapia o combinaciµn que se conocen por causar trombocitopenia grave. Las crisis de anemia de cÕlulas falciformes se asocian con la utilizaciµn de pegfilgrastim en pacientes con rasgo de cÕlulas falciformes o anemia de cÕlulas falciformes (ver Efectos colaterales). Por tanto, los mÕdicos deben tener precauciµn al prescribir Neulastim a pacientes con rasgo de cÕlulas falciformes o anemia de cÕlulas falciformes, se deben monitorizar los parÃmetros clÚnicos y de laboratorio y estar alerta sobre la posible asociaciµn de este medicamento con el aumento del tamaþo del bazo y una crisis vasooclusiva. Se han observado cuentas en los glµbulos blancos iguales o superiores a 100 x 109/l en menos del 1% de los pacientes tratados con Neulastim. No se han notificado eventos adversos directamente atribuibles a este grado de leucocitosis. Dichas elevaciones de los glµbulos blancos son pasajeras, normalmente ocurren de 24 a 48 horas despuÕs de la administraciµn y son consistentes con los efectos farmacodinÃmicos de este medicamento. En consistencia con los efectos clÚnicos y del potencial para leucocitosis, se deben realizar cuentas de leucocitos a intervalos regulares durante el tratamiento. Se debe interrumpir inmediatamente el tratamiento con este medicamento, si la cuenta de leucocitos supera los 50 x 109/l tras el punto mÚnimo esperado. Se ha notificado hipersensibilidad, incluyendo reacciones anafilÃcticas, que ocurrieron durante el tratamiento inicial o subsecuente en pacientes tratados con Neulastim. Suspenda definitivamente Neulastim en pacientes con hipersensibilidad clÚnica significativa. No administre Neulastim en pacientes con antecedentes de hipersensibilidad a pegfilgrastim o filgrastim. Ante una reacciµn alÕrgica grave, se debe administrar un tratamiento adecuado, con estrecho seguimiento del paciente durante varios dÚas. Como con todas las proteÚnas terapÕuticas, existe la posibilidad de inmunogenicidad. La tasa de generaciµn de anticuerpos contra pegfilgrastim es generalmente baja. Como con todos los biolµgicos, se generan anticuerpos de uniµn; sin embargo, hasta el momento no se han asociado con actividad neutralizante. La seguridad y eficacia de Neulastim para la movilizaciµn de cÕlulas progenitoras hematopoyÕticas en pacientes o donantes sanos no ha sido evaluada adecuadamente. La cubierta de la aguja de la jeringa prellenada contiene caucho natural (un derivado del lÃtex) que puede provocar reacciones alÕrgicas. El aumento de la actividad hematopoyÕtica en la mÕdula µsea en respuesta a la terapia con factores de crecimiento, se ha asociado con cambios transitorios de positividad µsea observables por imagen. Esto debe considerarse cuando se interpreten resultados de imÃgenes µseas. Efectos sobre la capacidad para conducir y utilizar maquinaria: La influencia de Neulastim sobre la capacidad para conducir y utilizar mÃquinas es nula o insignificante. Fertilidad: Pegfilgrastim no afectµ la capacidad reproductiva o la fertilidad en ratas hembras y machos a dosis semanales acumuladas de aproximadamente 6 a 9 veces mÃs elevadas de la dosis humana recomendada (basada en el Ãrea de superficie corporal). Embarazo: No existen o los datos sobre la utilizaciµn de pegfilgrastim en mujeres embarazadas son limitados. Estudios en animales han mostrado toxicidad reproductiva: No està recomendado el uso de Neulastim en mujeres embarazadas ni en mujeres en edad fÕrtil que no estÕn utilizando mÕtodos anticonceptivos. Lactancia: No existe informaciµn suficiente sobre la excreciµn de Neulastim/metabolitos en la leche materna. No se puede excluir un riesgo para los reciÕn nacidos/lactantes. Debe establecerse la decisiµn de suspender la lactancia o suspender/abstenerse de la terapia con Neulastim, teniendo en cuenta el beneficio de la lactancia para el niþo y el beneficio de la terapia para la mujer.
|
|
Interacciones Medicamentosas:
Debido a la potencial sensibilidad a la quimioterapia citotµxica de las cÕlulas mieloides en rÃpida divisiµn, Neulastim debe administrarse 24 horas despuÕs de la administraciµn de la quimioterapia citotµxica. En los estudios clÚnicos Neulastim se administrµ de forma segura 14 dÚas antes de la quimioterapia. La administraciµn simultÃnea de Neulastim con fÃrmacos quimioterapÕuticos no ha sido evaluada en pacientes. En modelos animales la administraciµn simultÃnea de Neulastim y 5 fluorouracilo (5-FU) u otros antimetabolitos ha mostrado potenciar la mielosupresiµn. En los ensayos clÚnicos no se ha investigado especÚficamente las posibles interacciones con otros factores de crecimiento hematopoyÕticos o con citocinas. No se ha investigado especÚficamente la posibilidad de interacciµn con el litio, que tambiÕn estimula la liberaciµn de los neutrµfilos. No hay evidencia de que dicha interacciµn sea nociva. La seguridad y eficacia de Neulastim no han sido evaluadas en pacientes tratados con fÃrmacos quimioterapÕuticos de acciµn mielosupresora retardada, p. ej. nitrosoureas. No se han realizados estudios especÚficos de interacciµn o metabolismo, sin embargo los ensayos clÚnicos no han indicado ninguna interacciµn entre Neulastim y cualquier otro medicamento.
|
|
Sobredosificaciµn:
Se han administrado por vÚa subcutÃnea dosis ºnicas de 300 çg/kg a un nºmero limitado de voluntarios sanos y a pacientes con cÃncer de pulmµn de cÕlulas no pequeþas sin eventos adversos graves. Los eventos adversos fueron similares a aquellos que se observaron en los pacientes que recibieron dosis menores de pegfilgrastim.
|
|
Incompatibilidades:
Este medicamento no debe mezclarse con otros medicamentos, especialmente con soluciones de cloruro sµdico.
|
|
Conservaciµn:
Almacenar a temperatura entre 2¯ C y 8¯ C. Neulastim puede dejarse a temperatura ambiente (que no supere los 30¤ C) durante un ºnico perÚodo de hasta 72 horas. Todo el Neulastim que haya permanecido a temperatura ambiente durante mÃs de 72 horas, debe desecharse. No congelar. La exposiciµn accidental a temperaturas de congelaciµn durante un perÚodo ºnico, inferior a 24 horas, no afecta la estabilidad de Neulastim. Conservar en el envase original para protegerlo de la luz.
|
|
Presentaciones:
Caja conteniendo 1 jeringa prellenada de 6 mg/0.6 ml de Neulastim soluciµn inyectable. Antes de administrar, la soluciµn de Neulastim debe ser inspeccionada visualmente y estar libre de partÚculas. Solamente deben inyectarse las soluciones que sean transparentes e incoloras. La agitaciµn excesiva puede producir el agregamiento de pegfilgrastim, haciÕndolo biolµgicamente inactivo. Dejar que la jeringa prellenada alcance la temperatura ambiente antes de inyectarla.
|
|



{kind=link}