 |
Composición:
Categor 150 mg: Cada comprimido recubierto contiene: 150 mg de Capecitabina. Categor 500 mg: Cada comprimido recubierto contiene: 500 mg de Capecitabina. Listado de excipientes: Núcleo del comprimido: Lactosa Monohidrato, Celulosa Microcristalina, Croscarmelosa Sodica, Hipromelosa, Estearato de Magnesio. Recubrimiento del comprimido: Hipromelosa, Talco, Dióxido de Titanio, Óxido de Hierro Rojo.
|
|
Acción Terapéutica:
Grupo fármaco-terapéutico: Análogos de pirimidinas. Código ATC: L01BC06.
|
|
Indicaciones:
Categor está indicado para el tratamiento coadyuvante de pacientes con posterioridad a cirugía de cáncer de colon fase III (fase C Duke) (ver sección Propiedades farmacodinámicas), en pacientes quienes han sufrido resección completa del tumor primario. Categor está indicado para el tratamiento de cáncer colorrectal metastásico (ver sección Propiedades farmacodinámicas). Categor está indicado para tratamiento de primera línea de cáncer esofagogástrico avanzado o metastásico en combinación con epirubicina y oxiplatino o cisplatino (ver sección Propiedades farmacodinámicas). Categor en combinación con docetaxel (ver sección Propiedades farmacodinámicas) está indicado para el tratamiento de pacientes con cáncer mamario metastásico o localmente avanzado posterior a fracaso de la quimioterapia citotóxica. La terapia previa debe haber incluido una antraciclina. Categor también está indicado como monoterapia para el tratamiento de pacientes con cáncer mamario metastásico o localmente avanzado después del fracaso de quimioterapia con taxanos y antraciclina o bien en aquellos en que no está indicado proseguir la terapia con antraciclina adicional.
|
|
Propiedades:
Propiedades farmacodinámicas: Capecitabina es un carbamato de fluoropirimidina no citotóxico que actúa como un precursor por vía oral del grupo citotóxico 5-fluorouracilo (5-FU). Capecitabina es activada mediante varias etapas enzimáticas (ver sección Propiedades farmacocinéticas). La enzima involucrada en la conversión final a 5-FU, la timidina fosforilasa (ThyPase), se encuentra en tejidos tumorales pero también en tejidos normales, aunque por lo general a niveles menores. En modelos de xenoinjerto de cáncer humano, capecitabina demostró un efecto sinérgico en combinación con docetaxel, que podría estar relacionado con la regulación al alza de la timidina fosforilasa por docetaxel. Existe evidencia de que el metabolismo del 5-FU en la vía anabólica bloquea la reacción de metilación del ácido desoxiuridílico al ácido timidílico, interfiriendo de esa forma con la síntesis del ácido desoxirribonucleico (ADN). La incorporación del 5-FU también lleva a la inhibición del ARN y la síntesis de proteínas. Dado que el ADN y el ARN son esenciales para la división y el crecimiento celular, el efecto del 5-FU podría ser crear una deficiencia de timidina que provoca el crecimiento desequilibrado y la muerte de la célula. Los efectos de la carencia de ADN y ARN son más pronunciados en aquellas células que proliferan con mayor rapidez y metabolizan 5-FU a una mayor velocidad. Cáncer de colon y colorrectal: Monoterapia con capecitabina en terapia coadyuvante de cáncer de colon: Datos de un ensayo clínico fase III, multicéntrico, aleatorizado y controlado en pacientes con cáncer de colon fase III (Duke C) respaldan el uso de capecitabina para tratamiento coadyuvante de pacientes con cáncer de colon (estudio XACT; M66001). En este ensayo, 1987 pacientes fueron distribuidos al azar para tratamiento con capecitabina (1250 mg/m2 2 veces al día durante 2 semanas, seguido por un período de descanso de 1 semana y administrado como ciclos de 3 semanas durante 24 semanas) o bien 5-FU y leucovorin (régimen Clínica Mayo: 20 mg/m2 de leucovorin IV seguido por 425 mg/m2 bolo IV de 5-FU, en los días 1 a 5, cada 28 días durante 24 semanas). Capecitabina fue al menos equivalente a 5-FU/LV IV en sobrevida libre de enfermedad en la población conforme al protocolo (cociente de riesgo 0.92; IC 95% 0.80-1.06). En la población aleatorizada total, las pruebas para detectar diferencia de capecitabina vs. 5-FU/LV en sobrevida total y sobrevida libre de enfermedad mostraron cocientes de riesgo de 0.88 (IC 95% 0.77 - 1.01; p = 0.068) y 0.86 (IC 95% 0.74 - 1.01; p = 0.060), respectivamente. La mediana de seguimiento al momento del análisis fue de 6.9 ańos. En un análisis Cox multivariable y preplanificado, se demostró la superioridad de capecitabina comparada con 5-FU/LV en bolo. Los siguientes factores fueron pre-especificados en el plan de análisis estadístico para su inclusión en el modelo: edad, tiempo desde la cirugía a la aleatorización, sexo, niveles CEA en la línea basal, nódulos linfáticos a nivel basal y país. En la población aleatorizada total, capecitabina mostró ser superior a 5-FU/LV para sobrevida libre de enfermedad (cociente de riesgo 0.849; IC 95% 0.739 - 0.976; p = 0.0212), al igual que para sobrevida total (cociente de riesgo 0.828; IC 95% 0.705 - 0.971; p = 0.0203). Terapia combinada en tratamiento coadyuvante de cáncer de colon: Datos de un ensayo clínico de fase 3, multicéntrico, aleatorizado y controlado en pacientes con cáncer de colon fase III (Dukes C) respaldan el uso de capecitabina en combinación con oxaliplatino (XELOX) para el tratamiento coadyuvante de pacientes con cáncer de colon (estudio NO16968). En este ensayo, 944 pacientes fueron distribuidos al azar a ciclos de 3 semanas durante 24 semanas con capecitabina (1000 mg/m2 2 veces al día durante 2 semanas, seguido por un período de descanso de 1 semana) en combinación con oxaliplatino (130 mg/m2 infusión intravenosa durante 2 horas el día 1 cada 3 semanas); 942 pacientes fueron aleatorizados a 5-FU y leucovorin en bolo. En el análisis primario para DFS en la población ITT, XELOX mostró ser significativamente superior a 5-FU/LV (HR=0.80, IC 95% =[0.69; 0.93]; p=0.0045). El porcentaje DFS a 3 ańos fue 71% para XELOX versus 67% para 5-FU/LV. El análisis de la variable secundaria de RFS respalda estos resultados, con un HR de 0.78 (IC 95% =[0.67; 0.92]; p=0.0024) para XELOX vs. 5-FU/LV. XELOX mostró una tendencia hacia un OS superior, con un HR de 0.87 (IC 95% =[0.72; 1.05]; p=0.1486) lo cual se traduce en un 13% de reducción en el riesgo de muerte. El porcentaje OS a 5 ańos fue de 78% para XELOX versus 74% para 5-FU/LV. Los datos de eficacia se basan en una mediana de tiempo de observación de 59 meses para OS y 57 meses para DFS. El porcentaje de discontinuación debido a eventos adversos fue mayor en el grupo de terapia combinada XELOX (21%) en comparación con el del grupo 5-FU/LV monoterapia (9%) en la población ITT. Monoterapia con capecitabina en cáncer colorrectal metastásico: Datos de 2 ensayos clínicos de fase III multicéntricos, aleatorizados y controlados de diseńo idéntico (SO14695; SO14796) respaldan el uso de capecitabina para tratamiento de primera línea de cáncer colorrectal metastásico. En estos ensayos, 603 pacientes fueron distribuidos al azar a tratamiento con capecitabina (1250 mg/m2 2 veces al día por 2 semanas, seguido por un período de descanso de 1 semana y administrado como ciclos de 3 semanas). 604 pacientes fueron aleatorizados a tratamiento con 5-FU y leucovorin (régimen Mayo: 20 mg/m2 leucovorin IV seguido por 425 mg/m2 IV bolo 5-FU, en los días 1 a 5, cada 28 días). Los porcentajes de respuesta objetiva totales en toda la población aleatorizada (evaluación del investigador) fueron 25.7% (capecitabina) vs. 16.7% (régimen Mayo); p <0.0002. La mediana de tiempo para progresión fue de 140 días (capecitabina) vs. 144 días (régimen Mayo). La mediana de sobrevida fue de 392 días (capecitabina) vs. 391 días (régimen Mayo). En la actualidad no se dispone de datos comparativos sobre capecitabina monoterapia en cáncer colorrectal comparado con regímenes asociados de primera línea. Terapia combinada en tratamiento de primera línea de cáncer colorrectal metastásico: Datos provenientes de un estudio clínico fase III, multicéntrico, aleatorizado y controlado (NO16966) respaldan el uso de capecitabina en combinación con oxaliplatino o bien asociado a oxaliplatino y bevacizumab para el tratamiento de primera línea de cáncer colorrectal metastásico. El estudio contenía 2 partes: una parte inicial de 2 grupos en la cual 634 pacientes fueron aleatorizados a 2 grupos terapéuticos distintos, incluyendo XELOX o FOLFOX-4, y una parte factorial 2x2 posterior en la cual 1401 pacientes fueron aleatorizados a 4 grupos terapéuticos distintos, incluyendo XELOX más placebo, FOLFOX-4 más placebo, XELOX más bevacizumab y FOLFOX-4 más bevacizumab. Ver la Tabla 6 para los regímenes terapéuticos.
Ver Tabla
La no inferioridad de los grupos conteniendo XELOX en comparación con los grupos conteniendo FOLFOX-4 en la comparación total fue demostrada en términos de sobrevida libre de progresión en la población de pacientes elegibles y la población con intención de tratar (ver Tabla 7). Los resultados indican que XELOX es equivalente a FOLFOX-4 en términos de sobrevida total (ver Tabla 7). Una comparación de XELOX más bevacizumab vs. FOLFOX-4 más bevacizumab fue un análisis exploratorio preespecificado. En esta comparación por subgrupos de tratamiento, XELOX más bevacizumab fue similar comparado a FOLFOX-4 más bevacizumab en términos de sobrevida libre de progresión (cociente de riesgo 1.01; IC 97.5% 0.84 - 1.22]). La mediana de seguimiento al momento de los análisis primarios en la población con intención de tratar fue de 1.5 ańos; los datos de los análisis después de 1 ańo de seguimiento adicional también se incluyen en la Tabla 7. Sin embargo, el análisis PFS con tratamiento no confirmó los resultados del PFS general y el análisis OS: el cociente de riesgo de XELOX vs. FOLFOX-4 fue de 1.24, con IC 97.5% 1.07 - 1.44. Aunque los análisis de sensibilidad muestran que las diferencias en los esquemas terapéuticos y el momento de las evaluaciones tumorales impactan en el análisis PFS con tratamiento, no se ha encontrado una explicación completa para este resultado.
Ver Tabla
Datos de un estudio fase III controlado y aleatorizado (CAIRO) respaldan el uso de capecitabina a una dosis inicial de 1000 mg/m2 por 2 semanas cada 3 semanas en combinación con irinotecan para el tratamiento de primera línea de pacientes con cáncer colorrectal metastásico. 820 pacientes fueron aleatorizados para recibir ya sea un tratamiento secuencial (n= 410) o tratamiento combinado (n= 410). El tratamiento secuencial consistió en un tratamiento de primera línea con capecitabina (1850 mg/m2 2 veces al día por 14 días), irinotecan de segunda línea (350 mg/m2 el día 1) y una combinación de capecitabina de tercera línea (1000 mg/m2 2 veces al día por 14 días) con oxaliplatino (130 mg/m2 el día 1). El tratamiento combinado consistió en un tratamiento de primera línea de capecitabina (1000 mg/m2 2 veces al día por 14 días) combinado con irinotecan (250 mg/m2 el día 1) (XELIRI) y capecitabina en segunda línea (1000 mg/m2 2 veces al día por 14 días) más oxaliplatino (130 mg/m2 el día 1). Todos los ciclos terapéuticos fueron administrados a intervalos de 3 semanas. En el tratamiento de primera línea, la mediana de sobrevida libre de progresión en la población con intención de tratar fue de 5.8 meses (IC 95% 5.1 - 6.2 meses) para capecitabina monoterapia y 7.8 meses (IC 95% 7.0 - 8.3 meses; p= 0.0002) para XELIRI. Datos de un análisis parcial de un estudio multicéntrico de fase II, aleatorizado y controlado (AIO KRK 0604) respaldan el uso de capecitabina a una dosis inicial de 800 mg/m2 por 2 semanas cada 3 semanas en combinación con irinotecan y bevacizumab para el tratamiento de primera línea de pacientes con cáncer colorrectal metastásico. 115 pacientes fueron aleatorizados a tratamiento con capecitabina combinada con irinotecan (XELIRI) y bevacizumab: capecitabina (800 mg/m2 2 veces al día durante 2 semanas, seguido por un período de descanso de 7 días), irinotecan (200 mg/m2 como infusión de 30 minutos el día 1 cada 3 semanas), un total de 118 pacientes fueron aleatorizados a tratamiento con capecitabina combinada con oxaliplatino más bevacizumab: capecitabina (1000 mg/m2 2 veces al día durante 2 semanas, seguido por un período de descanso de 7 días), oxaliplatino (130 mg/m2 como infusión de 2 horas el día 1 cada 3 semanas). La sobrevida libre de progresión a los 6 meses en la población con intención de tratar fue de 80% (XELIRI más bevacizumab) versus 74% (XELOX más bevacizumab). El porcentaje de respuesta total (respuesta completa más respuesta parcial) fue de 45% (XELOX más bevacizumab) versus 47% (XELIRI más bevacizumab). Terapia combinada en tratamiento de segunda línea de cáncer colorrectal metastásico: Datos de un estudio clínico fase III multicéntrico, aleatorizado y controlado (NO16967) respaldan el uso de capecitabina en combinación con oxaliplatino para el tratamiento de segunda línea de cáncer colorrectal metastásico. En este ensayo, 627 pacientes con carcinoma colorrectal metastásico que han recibido tratamiento previo con irinotecan en combinación con una fluoropirimidina como terapia de primera línea fueron aleatorizados a tratamiento con XELOX o FOLFOX-4. Para el esquema de dosificación de XELOX y FOLFOX-4 (sin adición de placebo o bevacizumab), consultar la Tabla 6. XELOX demostró ser no inferior a FOLFOX-4 en términos de sobrevida libre de progresión en la población conforme al protocolo y la población con intención de tratar (ver Tabla 8). Los resultados indican que XELOX es equivalente a FOLFOX-4 en términos de sobrevida total (ver Tabla 8). La mediana de seguimiento al momento de los análisis primarios en la población con intención de tratar fue de 2.1 ańos; los datos de los análisis posteriores a 6 meses adicionales de seguimiento también se incluyen en la Tabla 8.
Ver Tabla
Cáncer gástrico avanzado: Datos de un ensayo clínico de fase III, multicéntrico, aleatorizado y controlado en pacientes con cáncer gástrico avanzado respaldan el uso de capecitabina para el tratamiento de primera línea de cáncer gástrico avanzado (ML17032). En este ensayo, 160 pacientes fueron aleatorizados a tratamiento con capecitabina (1000 mg/m2 2 veces al día durante 2 semanas, seguido por un período de descanso de 7 días) y cisplatino (80 mg/m2 como infusión de 2 horas cada 3 semanas). Un total de 156 pacientes fueron aleatorizados a tratamiento con 5-FU (800 mg/m2 al día, infusión continua los días 1 a 5 cada 3 semanas) y cisplatino (80 mg/m2 como infusión de 2 horas el día 1 cada 3 semanas). Capecitabina en combinación con cisplatino fue no inferior a 5-FU en combinación con cisplatino en términos de sobrevida libre de progresión en el análisis conforme al protocolo (cociente de riesgo 0.81; IC 95% 0.63 -1.04). La mediana de la sobrevida libre de progresión fue de 5.6 meses (capecitabina + cisplatino) versus 5.0 meses (5-FU + cisplatino). El cociente de riesgo para la duración de la sobrevida (sobrevida total) fue similar al cociente de riesgo para la sobrevida libre de progresión (cociente de riesgo 0.85; IC 95% 0.64 -1.13). La duración mediana de la sobrevida fue de 10.5 meses (capecitabina + cisplatino) versus 9.3 meses (5-FU + cisplatino). Datos de un estudio de fase III multicéntrico y aleatorizado que comparó capecitabina con 5-FU y oxaliplatino con cisplatino en pacientes con cáncer gástrico avanzado respaldan el uso de capecitabina para tratamiento de primera línea de cáncer gástrico avanzado (REAL-2). En este ensayo, 1002 pacientes fueron aleatorizados en un diseńo factorial 2x2 a uno de los siguientes 4 grupos: ECF: epirrubicina (50 mg/ m2 como bolo el día 1 cada 3 semanas), cisplatino (60 mg/m2 como infusión de 2 horas el día 1 cada 3 semanas) y 5-FU (200 mg/m2 diarios administrados como infusión continua mediante una línea central). ECX: epirrubicina (50 mg/m2 como bolo el día 1 cada 3 semanas), cisplatino (60 mg/m2 como infusión de 2 horas el día 1 cada 3 semanas) y capecitabina (625 mg/m2 2 veces al día en forma continua). EOF: epirrubicina (50 mg/m2 como bolo el día 1 cada 3 semanas), oxaliplatino (130 mg/m2 administrado como infusión de 2 horas el día 1 cada 3 semanas) y 5-FU (200 mg/m2 diarios administrados por infusión continua mediante una línea central). EOX: epirrubicina (50 mg/m2 como bolo el día 1 cada 3 semanas), oxaliplatino (130 mg/m2 administrado como infusión de 2 horas el día 1 cada 3 semanas) y capecitabina (625 mg/m2 2 veces al día en forma continua). Los análisis de eficacia primaria en la población conforme al protocolo demostraron no inferioridad en sobrevida total para regímenes con capecitabina vs. regímenes con 5-FU (cociente de riesgo 0.86; IC 95% 0.8-0.99) y para regímenes a base de oxaliplatino vs. cisplatino (cociente de riesgo 0.92; IC 95% 0.80-1.1). La mediana de sobrevida total fue de 10.9 meses en regímenes con capecitabina y 9.6 meses en regímenes con 5-FU. La mediana de sobrevida total fue de 10.0 meses en regímenes con cisplatino y 10.4 meses en regímenes con oxaliplatino. Capecitabina también se ha utilizado en combinación con oxaliplatino para el tratamiento de cáncer gástrico avanzado. Estudios con capecitabina monoterapia indican que capecitabina tiene actividad en cáncer gástrico avanzado. Cáncer de colon, cáncer colorrectal y cáncer gástrico avanzado: metaanálisis. Un metaanálisis de 6 ensayos clínicos (estudios SO14695, SO14796, M66001, NO16966, NO16967, M17032) respalda el reemplazo de capecitabina por 5-FU para monoterapia y tratamiento combinado en cáncer gastrointestinal. El análisis combinado incluye 3097 pacientes tratados con regímenes que contienen capecitabina y 3074 pacientes tratados con regímenes que contienen 5-FU. La mediana de tiempo de sobrevida total fue de 703 días (IC 95%: 671; 745) en pacientes tratados con regímenes con capecitabina y 683 días (IC 95%: 646; 715) en pacientes tratados con regímenes con 5-FU. El cociente de riesgo para sobrevida total fue de 0.94 (IC 95%: 0.89; 1.00, p= 0.0489), lo que indica que los regímenes con capecitabina son superiores a los regímenes que contienen 5-FU. Cáncer de mama: Terapia combinada con capecitabina y docetaxel en cáncer mamario metastásico o localmente avanzado: Datos de un ensayo clínico multicéntrico, aleatorizado y controlado de fase III respaldan el uso de capecitabina en combinación con docetaxel para el tratamiento de pacientes con cáncer mamario metastásico o localmente avanzado después del fracaso de quimioterapia citotóxica, incluyendo una antraciclina. En este ensayo, 255 pacientes fueron aleatorizados a tratamiento con capecitabina (1250 mg/m2 2 veces al día durante 2 semanas, seguido por un período de descanso de 1 semana y docetaxel 75 mg/m2 como infusión intravenosa de 1 hora cada 3 semanas). 256 pacientes fueron aleatorizados a tratamiento con docetaxel solo (100 mg/m2 como infusión intravenosa de 1 hora cada 3 semanas). La sobrevida fue superior en el grupo con la asociación capecitabina+ docetaxel (p= 0.0126). La sobrevida mediana fue de 442 días (capecitabina+ docetaxel) vs. 352 días (docetaxel solo). Los porcentajes objetivos totales en toda la población aleatorizada (evaluación del investigador) fueron 41.6% (capecitabina + docetaxel) vs. 29.7% (docetaxel solo); p = 0.0058. El tiempo hasta enfermedad progresiva fue superior en el brazo con la asociación capecitabina+ docetaxel (p<0.0001). La mediana de tiempo para progresión fue de 186 días (capecitabina + docetaxel) vs. 128 días (docetaxel solo). Monoterapia con capecitabina después del fracaso de taxanos, quimioterapia conteniendo antraciclina y para quienes no está indicada la terapia con antraciclina: Datos provenientes de 2 ensayos clínicos multicéntricos de fase II respaldan el uso de capecitabina monoterapia para el tratamiento de pacientes después del fracaso de taxanos y un régimen de quimioterapia conteniendo antraciclina, o bien para quienes la terapia con antraciclina adicional no está indicada. En estos ensayos, un total de 236 pacientes fueron tratados con capecitabina (1250 mg/m2 2 veces al día durante 2 semanas o más seguido por un período de descanso de 1 semana). Los porcentajes de respuesta objetiva total (evaluación del investigador) fueron de 20% (primer ensayo) y 25% (segundo ensayo). La mediana del tiempo hasta la progresión fue de 93 y 98 días. La sobrevida mediana fue de 384 y 373 días. Indicaciones totales: Un metaanálisis de 14 ensayos clínicos con datos de más de 4700 pacientes tratados con capecitabina monoterapia o capecitabina asociada a diferentes regímenes de quimioterapia en indicaciones múltiples (cáncer de colon, colorrectal, gástrico y mamario), mostró que los pacientes con capecitabina que desarrollaron síndrome mano-pie (HFS) tuvieron una mayor sobrevida total comparado con pacientes que no desarrollaron HFS: mediana de sobrevida total de 1100 días (IC 95% 1007; 1200) vs 691 días (IC 95% 638; 754) con un cociente de riesgo de 0.61 (IC 95% 0.56; 0.66). Propiedades farmacocinéticas: La farmacocinética de capecitabina ha sido evaluada respecto a un rango de dosis de 502 - 3514 mg/m2/día. Los parámetros de capecitabina, 5'-desoxi-5-fluorocitidina (5'-DFCR) y 5'-desoxi-5-fluorouridina (5'-DFUR) medidos en los días 1 y 14 fueron similares. El AUC de 5-FU fue un 30% -35% mayor el día 14. La reducción de dosis de capecitabina disminuye la exposición sistémica a 5-FU más que la proporcionalidad de dosis debido a la farmacocinética no lineal del metabolito activo. Absorción: después de la administración oral, capecitabina es absorbida en forma rápida y extensa, seguido por una amplia conversión a los metabolitos 5'-DFCR y 5'-DFUR. La administración con alimentos disminuye el porcentaje de absorción de capecitabina, pero sólo se traduce en un efecto menor en el AUC de 5'-DFUR, y en el AUC del metabolito posterior 5-FU. A la dosis de 1250 mg/m2 el día 14 con una administración posterior a la ingesta de alimentos, las concentraciones plasmáticas máximas (Cmax en µg/ml) para capecitabina, 5'-DFCR, 5'-DFUR, 5-FU y FBAL fueron 4.67, 3.05, 12.1, 0.95 y 5.46 respectivamente. El tiempo para las concentraciones plasmáticas máximas (Tmax en horas) fueron 1.50, 2.00, 2.00, 2.00 y 3.34. Los valores AUC0-Ą en µg · h/ml fueron 7.75, 7.24, 24.6, 2.03 y 36.3. Unión a proteínas: estudios en plasma humano in vitro han establecido que capecitabina, 5'-DFCR, 5'-DFUR y 5-FU se unen un 54%, 10%, 62% y 10% a proteínas, principalmente a albúmina. Metabolismo: capecitabina es metabolizada primero por la carboxilesterasa hepática a 5'-DFCR, el cual es luego convertido a 5'-DFUR por la citidina desaminasa, localizada principalmente en el hígado y tejidos tumorales. A continuación ocurre la activación catalítica adicional de 5'-DFUR por la timidina fosforilasa (ThyPase). Las enzimas involucradas en la activación catalítica se encuentran en tejidos tumorales pero también en tejidos normales, aunque habitualmente a niveles menores. La biotransformación enzimática secuencial de capecitabina a 5-FU lleva a mayores concentraciones dentro de los tejidos tumorales. En el caso de tumores colorrectales, la generación de 5-FU parece ocurrir en gran parte en las células estromales tumorales. Después de la administración oral de capecitabina a pacientes con cáncer colorrectal, la proporción de concentración de 5-FU en tumores colorrectales respecto a tejidos adyacentes fue de 3.2 (variación de 0.9 a 8.0). La proporción de concentración de 5-FU en tumor respecto a plasma fue de 21.4 (variación de 3.9 a 59.9, n=8), mientras que la proporción en tejidos sanos respecto a plasma fue de 8.9 (variación de 3.0 a 25.8, n=8). Se midió la actividad de la timidina fosforilasa y resultó ser 4 veces mayor en tumor colorrectal primario que en tejido normal adyacente. De acuerdo a estudios inmunohistoquímicos, la timidina fosforilasa parece estar en gran parte localizada en las células estromales tumorales. El 5-FU vuelve a ser catabolizado por la enzima dihidropirimidina deshidrogenasa (DPD) a la molécula mucho menos tóxica dihidro-5-fluorouracilo (FUH2). La dihidropirimidinasa descompone el anillo de pirimidina para obtener el ácido 5-fluoro-ureidopropiónico (FUPA). Finalmente, la ß-ureido-propionasa degrada el FUPA a a -fluoro-ß-alanina (FBAL), la cual es eliminada en la orina. La actividad de la dihidropirimidina deshidrogenasa (DPD) es la etapa limitadora de velocidad. La deficiencia de DPD puede llevar a una mayor toxicidad de capecitabina (ver sección Contraindicaciones y Advertencias especiales y precauciones especiales para el uso). Eliminación: la vida media de eliminación (t1/2 en horas) de capecitabina, 5'-DFCR, 5'-DFUR, 5-FU y FBAL fueron 0.85, 1.11, 0.66, 0.76 y 3.23 respectivamente. Capecitabina y sus metabolitos son excretados principalmente en la orina; el 95.5% de la dosis de capecitabina administrada es recuperada en la orina. La excreción fecal es mínima (2.6%). El principal metabolito excretado en la orina es FBAL, el cual representa el 57% de la dosis administrada. Cerca del 3% de la dosis administrada es excretada en la orina como fármaco inalterado. Terapia combinada: estudios de fase I que evaluaron el efecto de capecitabina en la farmacocinética de docetaxel o paclitaxel y viceversa no mostraron un efecto de capecitabina sobre la farmacocinética de docetaxel o paclitaxel (Cmax y AUC) y tampoco de docetaxel o paclitaxel sobre la farmacocinética de 5'-DFUR. Farmacocinética en poblaciones especiales: Se realizó un análisis farmacocinético de poblaciones después de tratamiento con capecitabina en 505 pacientes con cáncer colorrectal a la dosis de 1850 mg/m2 2 veces al día. El sexo, la presencia o la ausencia de metástasis hepática a nivel basal, escala de rendimiento de Karnofsky, bilirrubina total, albúmina en suero, ASAT y ALAT no tuvieron un efecto estadísticamente significativo en la farmacocinética de 5'-DFUR, 5-FU y FBAL. Pacientes con dańo hepático debido a metástasis hepática: De acuerdo a un estudio farmacocinético en pacientes con cáncer con dańo hepático leve a moderado debido a metástasis hepática, la biodisponibilidad de capecitabina y la exposición a 5-FU podrían aumentar respecto a pacientes sin dańo hepático. No existen datos farmacocinéticos sobre pacientes con dańo hepático severo. Pacientes con dańo renal: En base a un estudio farmacocinético en pacientes con cáncer con dańo renal leve a severo, no existe evidencia de algún efecto sobre la depuración de creatinina en la farmacocinética del fármaco intacto y 5-FU. Se encontró que la depuración de creatinina influye en la exposición sistémica a 5'-DFUR (35% de aumento de AUC cuando la depuración de creatinina disminuye en un 50%) y a FBAL (114% de aumento en el AUC cuando la depuración de creatinina disminuye en un 50%). FBAL es un metabolito sin actividad antiproliferativa. Ancianos: En base al análisis farmacocinético poblacional, que incluyó pacientes con un amplio rango de edad (27 a 86 ańos) e incluyó 234 pacientes (46%) de 65 ańos en adelante, la edad no influye en la farmacocinética de 5'-DFUR y 5-FU. El AUC de FBAL aumentó con la edad (20% de incremento en la edad lleva a un 15% de aumento en el AUC de FBAL). Este aumento se debe probablemente a un cambio en la función renal. Factores étnicos: Después de la administración oral de 825 mg/m2 de capecitabina 2 veces al día por 14 días, los pacientes japoneses (n=18) tuvieron un Cmax 36% menor y un AUC 24% menor para capecitabina respecto a pacientes caucásicos (n=22). Los pacientes japoneses tuvieron además un Cmax 25% menor y un AUC 34% menor para FBAL que los pacientes caucásicos. La relevancia clínica de estas diferencias es desconocida. No hubo diferencias significativas en la exposición a otros metabolitos (5'-DFCR, 5'-DFUR y 5-FU). Datos de seguridad preclínicos: En estudios de toxicidad con dosis repetidas, la administración oral diaria de capecitabina a monos cinomolgos y ratones causó efectos tóxicos a nivel de sistema gastrointestinal, linfático y hematopoyético, típico de las fluoropirimidinas. Estas toxicidades fueron reversibles. Se observó toxicidad cutánea, caracterizada por cambios degenerativos/regresivos, con capecitabina. Capecitabina estuvo desprovista de toxicidad hepática y sobre el SNC. La toxicidad cardiovascular (por ejemplo, prolongación de intervalo PR y QT) fue detectable en monos cinomolgos después de la administración intravenosa (100 mg/kg) pero no después de dosificación oral repetida (1379 mg/m2/día). Un estudio de carcinogenicidad de 2 ańos en ratones no arrojó evidencia de carcinogenicidad por capecitabina. Durante estudios de fertilidad estándar, se observó alteración de la fertilidad en ratones hembras que recibieron capecitabina; sin embargo, este efecto fue reversible después de un período libre de fármaco. Además, durante un estudio de 13 semanas, ocurrieron cambios degenerativos y atróficos en órganos reproductivos de ratones machos; sin embargo estos efectos fueron reversibles después de un período libre de fármaco. En estudios de teratogenicidad y toxicidad embrionaria en ratones, se observaron incrementos dosis dependientes en resorción fetal y teratogenicidad. En monos se observó aborto y letalidad embrionaria en dosis elevadas, pero no hubo evidencia de teratogenicidad. Capecitabina no fue mutagénica in vitro para bacterias (prueba Ames) o células de mamíferos (ensayo de mutaciones genéticas en hámster chino V79/HPRT). Sin embargo, en forma similar a otros análogos de nucleósido (es decir, 5-FU), capecitabina fue clastogénica en linfocitos humanos (in vitro) y hubo una tendencia positiva en ensayos de micronúcleos de médula ósea de ratón (in vivo).
|
|
Posología:
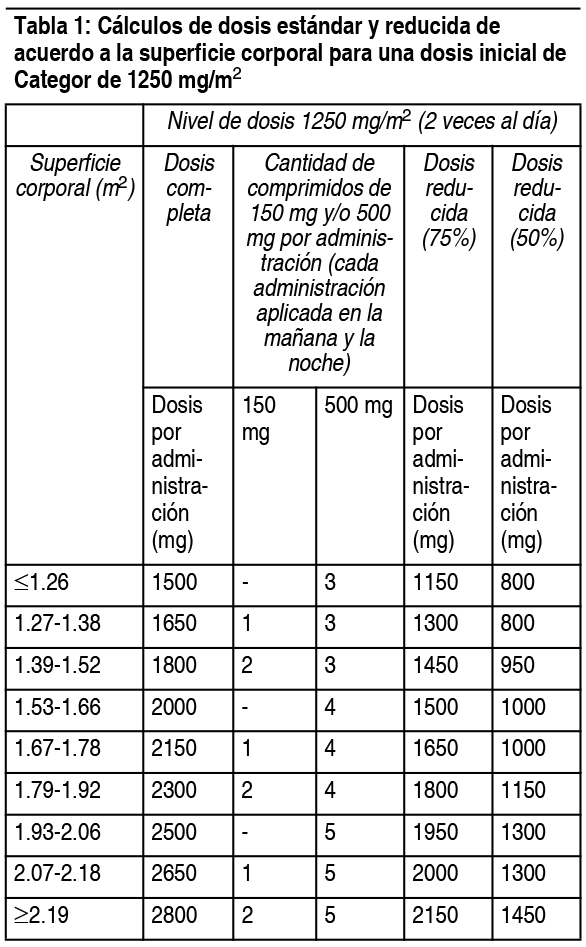
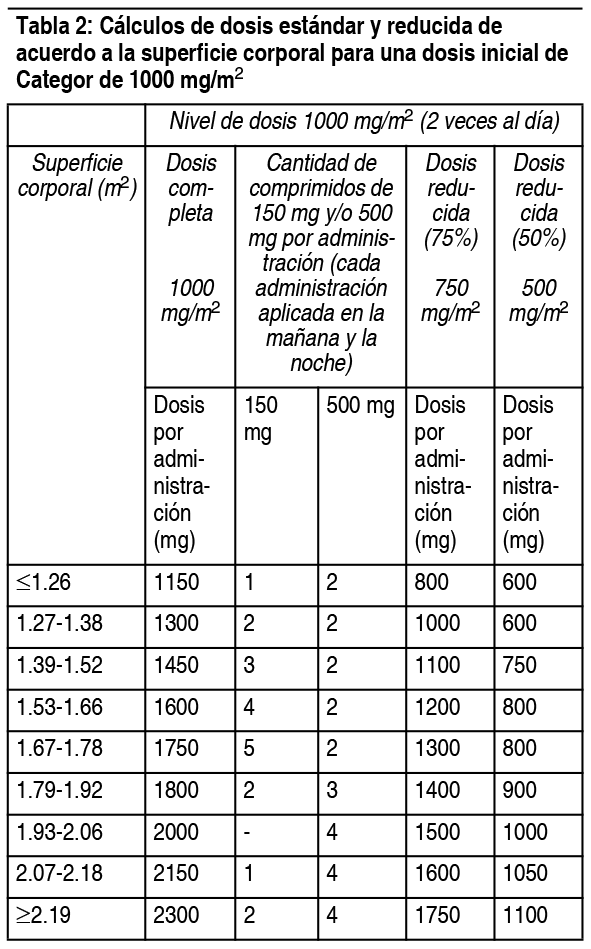
Categor sólo debe ser indicado por un médico calificado con experiencia en la utilización de agentes antineoplásicos. Los comprimidos de Categor se deben tragar con agua dentro de los 30 minutos posteriores a una comida. El tratamiento debe ser discontinuado si se observa enfermedad progresiva o toxicidad intolerable. Los cálculos de dosis estándar y reducidas de acuerdo a la superficie corporal para las dosis iniciales de Categor de 1250 mg/m2 y 1000 mg/m2 se entregan en las tablas 1 y 2, respectivamente. Posología recomendada (ver sección Propiedades farmacodinámicas): Monoterapia: Administrado como agente único, la dosis inicial recomendada para Categor es de 1250 mg/m2 administrados 2 veces al día (mańana y noche; equivalente a 2500 mg/m2 dosis diaria total) durante 14 días, seguido por un período de descanso de 7 días. Categor cálculos de dosis:
Ver Tabla
Ver Tabla
En asociación con docetaxel: En asociación con docetaxel, la dosis recomendada de capecitabina es 1250 mg/m2 2 veces al día durante 2 semanas, seguido de 7 dias de reposo farmacológico y la de docetaxel de 75 mg/m2 en infusión intravenosa (I.V.) de 1 hora, cada 3 semanas. De acuerdo con la información sobre docetaxel en los pacientes tratados con capecitabina junto con docetaxel debe iniciarse una premedicación antes de administrarse el docetaxel. En asociación con cisplatino: En asociación con cisplatino, la dosis recomendada de capecitabina e de 1000 mg/m2 2 veces al día durante 2 semanas, seguido de 7 días de reposo farmacológico. El cisplatino se administra en una dosis de 80 mg/m2 en infusión I.V. de 2 horas el día 1 de cada 3 semanas. La primera dosis de capecitabina se administra por la noche en día 1 y la última dosis por la mańana el día 15. DE acuerdo con la información sobre cisplatino, en los pacientes tratados con capecitabina junto con cisplatino debe iniciarse premedicación antiemética y suficientemente hidratante antes de administrarse el cisplatino. En asociación con oxaliplatino y/o bevacizumab: En asociación con oxiliplatino y/o bevacizumab, la dosis recomendada de capecitabina es de 1000 mg/m2 2 veces al día durante 2 semanas, seguido de 7 días de reposo farmacológico. La primera dosis de capecitabina se administra en la víspera del día 1 y la última dosis por la mańana de día 15. En una pauta de 1 vez (el día 1) cada 3 semanas, el bevacizumab se administra en infusión I.V. de 7.5 mg/kg durante 30 a 90 minutos, seguido de oxaliplatino en infusión I.V. de 7.5 mg/kg durante 2 horas. En asociación con oxaliplatino y epirrubicina: La dosis recomendad de capecitabina es de 625 mg/m2 2 veces al día durante 24 semanas sin interrupción en combinación con 130 mg/m2 (por infusión intravenosa de 2 o más horas) de oxaliplatino (cada 3 semanas, máximo 8 ciclos) y 50 mg/m2 de epirrubicina por bolo I.V. (cada 3 semanas, máximo 8 ciclos). Para obtener más información de la premedicacion para mantener la hidratación adecuada y el tratamiento antiemético antes de la administración de oxaliplatino, referirse a la información disponible para ese producto. En asociación con cisplatino y epirrubicina: La dosis recomendada de capecitabina es de 625 mg/m2 2 veces al día durante 24 semanas sin interrupción, en combinación con 60 mg/m2 de cisplatino (cada 3 semanas, máximo 8 ciclos) y 50 mg/m2 de epirrubicina por bolo I.V. (cada 3 semanas, máximo 8 ciclos). De acuerdo con la información sobre cisplatino, en los pacientes tratados con capecitabina junto con cisplatino debe iniciarse premedicación antiemética y suficientemente hidratante antes de administrar el cisplatino. Ajustes de dosis durante el tratamiento: Aspectos generales: La toxicidad por administración de Categor se puede manejar con tratamiento sintomático y/o modificación de la dosis (interrupción del tratamiento o reducción de la dosis). Una vez que la dosis ha sido reducida, no se debería aumentar con posterioridad. Para aquellas toxicidades que el médico tratante considera que probablemente no serán un riesgo grave o vital, como la alopecia, alteración del sabor, cambios en las uńas, el tratamiento se puede continuar a la misma dosis sin su disminución o interrupción. Los pacientes que toman Categor deben ser informados de la necesidad de interrumpir el tratamiento en forma inmediata si ocurre toxicidad moderada o severa. Las dosis de Categor omitidas por toxicidad no son reemplazadas. Las siguientes son las modificaciones de dosis recomendadas para toxicidad:
Ver Tabla
Hematología: Pacientes con conteo basal de neutrófilos <1.5 x 109/l y/o conteo de trombocitos <100 x 109/l no deben ser tratados con Categor. Si las evaluaciones de laboratorio no programadas durante un ciclo terapéutico muestran que el conteo de neutrófilos cae por debajo de 1.0 x 109/l o que el conteo de plaquetas cae por debajo de 75 x 109/l, el tratamiento con Categor debe ser interrumpido. Modificaciones de dosis por toxicidad cuando Categor se utiliza como ciclo de 3 semanas en combinación con otros agentes: Las modificaciones de dosis por toxicidad cuando Categor se utiliza por 3 semanas en combinación con otros agentes deben ser efectuadas de acuerdo a la Tabla 3 anterior para Categor y de acuerdo al resumen de características del producto correspondiente para los otros agentes. Al inicio de un ciclo de tratamiento, si se indica retardo del tratamiento para Categor o el otro agente, entonces la administración de todos los agentes se debe retardar hasta que se cumplan los requerimientos para reiniciar todos los fármacos. Durante un ciclo de tratamiento, para aquellas toxicidades consideradas por el médico tratante como no relacionadas con Categor, se debe continuar con Categor y la dosis del otro agente debe ser ajustada de acuerdo a la Información para Prescribir correspondiente. Si el otro agente tiene que ser discontinuado a permanencia, el tratamiento con Categor puede ser retomado cuando se cumplen los requerimientos para reiniciar Categor. Esta recomendación se aplica a todas las indicaciones y a todas las poblaciones especiales. Modificaciones de dosis por toxicidad cuando Categor se usa continuamente en combinación con otros agentes: Las modificaciones de dosis por toxicidad cuando Categor se usa continuamente en combinación con otros agentes se deben realizar de acuerdo a la Tabla 3 anterior para Categor y de acuerdo al resumen de características del producto que corresponda para el otro agente. Ajuste de dosis para poblaciones especiales: Dańo hepático: se dispone de información de eficacia y seguridad insuficiente en pacientes con dańo hepático para proporcionar una recomendación sobre ajuste de dosis. No se dispone de información sobre dańo hepático por cirrosis o hepatitis. Dańo renal: Categor está contraindicado en pacientes con dańo renal severo (depuración de creatinina menor a 30 ml/min [Cockcroft y Gault] a nivel basal). La incidencia de reacciones adversas grado 3 ó 4 en pacientes con dańo renal moderado (depuración de creatinina de 3050 ml/min a nivel basal) está aumentada en comparación con la población total. En pacientes con dańo renal moderado a nivel basal, se recomienda una reducción de dosis al 75% para una dosis inicial de 1250 mg/m2. En pacientes con dańo renal moderado a nivel basal, no se requiere reducción de dosis para una dosis inicial de 1000 mg/m2. En pacientes con dańo renal leve (depuración de creatinina de 5180 ml/min a nivel basal) no se recomienda ajustar la dosis inicial. Se recomienda un monitoreo cuidadoso y la pronta interrupción del tratamiento si el paciente desarrolla un evento adverso grado 2, 3 ó 4 durante el tratamiento y el posterior ajuste de dosis tal cual se destaca en la Tabla 3 anterior. Si la depuración de creatinina calculada disminuye durante el tratamiento hasta un valor inferior a 30 ml/min, Categor debe ser discontinuado. Estas recomendaciones de ajuste de dosis para dańo renal se aplican al uso como monoterapia y en asociación (ver también sección "Ancianos" a continuación). No existe experiencia en nińos (menores de 18 ańos). Ancianos: Durante monoterapia con Categor, no es necesario ajustar la dosis inicial. Sin embargo, las reacciones adversas grado 3 ó 4 asociados al tratamiento fueron más frecuentes en pacientes ł 60 ańos de edad en comparación con pacientes más jóvenes. Cuando se utilizó Categor asociado a otros agentes, los pacientes ancianos ( ł 65 ańos) experimentaron más reacciones farmacológicas adversas grado 3 y 4, incluyendo las que llevaron a la discontinuación, comparado con pacientes de menor edad. Se recomienda un monitoreo cuidadoso de pacientes >60 ańos de edad. En combinación con docetaxel: se observó una mayor incidencia de reacciones adversas grado 3 ó 4 y reacciones adversas graves asociadas al tratamiento en pacientes mayores de 60 ańos de edad (ver sección Propiedades farmacodinámicas). Para pacientes mayores de 60 ańos, se recomienda disminuir la dosis inicial de Categor al 75% (950 mg/m2 2 veces al día). Si no se observa toxicidad en pacientes >60 ańos de edad tratados con una dosis inicial reducida de Categor en combinación con docetaxel, la dosis de Categor se puede escalar con precaución hasta 1250 mg/m2 2 veces al día. En combinación con irinotecan: para pacientes mayores de 65 ańos de edad, se recomienda la reducción de la dosis inicial de capecitabina a 800 mg/m2 2 veces al día.
|
|
Efectos Colaterales:
Resumen del perfil de seguridad: El perfil de seguridad global de capecitabina se basa en datos provenientes de más de 3000 pacientes tratados con capecitabina como monoterapia o capecitabina asociada a distintos regímenes de quimioterapia en múltiples indicaciones. Los perfiles de seguridad de capecitabina monoterapia para poblaciones con cáncer mamario metastásico, cáncer colorrectal metastásico y cáncer de colon como coadyuvante son comparables. Ver sección Propiedades farmacodinámicas para detalles sobre los estudios principales, incluyendo diseńos y principales resultados de eficacia. Las reacciones farmacológicas adversas (ADRs) asociadas al tratamiento informadas con mayor frecuencia o bien clínicamente relevantes son los trastornos gastrointestinales (especialmente la diarrea, náuseas, vómito, dolor abdominal, estomatitis), síndrome mano-pie (eritrodisestesia palmar -plantar), fatiga, astenia, anorexia, cardiotoxicidad, mayor disfunción renal en personas con compromiso preexistente de la función renal y trombosis/embolismo. Resumen tabulado de reacciones adversas: Las ADRs consideradas por el investigador como posible, probable o remotamente relacionadas con la administración de capecitabina se detallan en la Lista 1 para capecitabina administrada como agente único y en la Lista 2 para capecitabina administrada en combinación con diferentes regímenes de quimioterapia en indicaciones múltiples. Se utiliza la siguiente nomenclatura para clasificar las ADRs por frecuencia: ( ł 1/10), común ( ł 1/100, < 1/10) y no común ( ł 1/1,000, < 1/100). Dentro de cada grupo de frecuencias, las ADRs se presentan en orden de gravedad decreciente. Capecitabina monoterapia: La lista 1 contiene las ADRs asociadas al uso de capecitabina monoterapia en base al análisis combinado de datos de seguridad de los 3 principales estudios que incluyen a más de 1900 pacientes (estudios M66001, SO14695 y SO14796). Las ADRs son agregadas al grupo de frecuencia correspondiente de acuerdo a la incidencia total a partir del análisis combinado. Lista 1: Resumen de ADS reportados en pacientes tratados con capecitabina como monoterapia. Infecciones e infestaciones: Comunes/Todos los grados: Infección por herpes viral, nasofaringitis, infección del tracto respiratorio inferior. Poco común / grave o potencialmente fatal (grado 3-4) o considerado médicamente relevante: Sepsis, infección del tracto urinario, celulitis, tonsilitis, faringitis, candidiasis oral, influenza, gastroenteritis, Infección por hongos, infección herpética, abscesos dental. Neoplasmas benignos, malignos e inespecíficos: Poco común / grave o potencialmente fatal (grado 3-4) o considerado médicamente relevante: Lipoma. Trastornos de la sangre y sistema linfático: Común / Todos los grados: Neutropenia, anemia. Poco común / grave o potencialmente fatal (grado 3-4) o considerado médicamente relevante: neutropenia febril, pancitopenia, granulocitopenia, trombocitopenia, leucopenia, anemia hemolítica, incremento en Cociente Internacional Normalizado (INR)/tiempo de protrombina prolongado. Trastornos del sistema inmunológico: Poco común / grave o potencialmente fatal (grado 3-4) o considerado médicamente relevante: Hipersensibilidad. Trastornos del metabolismo y nutrición: Muy común/todos los grados: Anorexia. Común / todos los grados: Deshidratación, menor apetito, disminución de peso. Poco común / grave o potencialmente fatal (grado 3-4) o considerado médicamente relevante: Diabetes, hipocalcemia, desórdenes del apetito, desnutrición, hipertrigliceridemia. Trastornos siquiátricos: Común / todos los grados: Insomnio, depresión. Poco común / grave o potencialmente fatal (grado 3-4) o considerado médicamente relevante: Estado de confusión, ataque de pánico, estado de ánimo deprimido, disminución en la libido. Trastornos del sistema nervioso: Común / todos los grados: cefalea, letargo, mareos, parestesia, disgeusia. Poco común / grave o potencialmente fatal (grado 3-4) o considerado médicamente relevante: Afasia, deterioro de la memoria, ataxia, síncope, alteraciones del equilibrio, trastorno sensorial, neuropatía periférica. Trastornos del ojo: Común / todos los grados: incremento en lagrimeo, conjuntivitis, irritación ocular. Poco común / grave o potencialmente fatal (grado 3-4) o considerado médicamente relevante: reducción de agudeza visual, diplopía. Trastornos del oído y laberinto: Poco común / grave o potencialmente fatal (grado 3-4) o considerado médicamente relevante: Vértigo, dolor de oído. Trastornos cardíacos: Poco común / grave o potencialmente fatal (grado 3-4) o considerado médicamente relevante: Angina inestable, angina pectoris, isquemia del miocardio, fibrilación auricular, arritmia, taquicardia, taquicardia sinusal, palpitaciones. Trastornos vasculares: Común / todos los grados: Tromboflebitis. Poco común / grave o potencialmente fatal (grado 3-4) o considerado médicamente relevante: Trombosis venosa profunda, hipertensión, petequias, hipotensión, rubores, enfriamiento periférico. Trastornos respiratorios, torácicos y del mediastino: Común / todos los grados: Disnea, epistaxis, tos, rinorrea. Poco común / grave o potencialmente fatal (grado 3-4) o considerado médicamente relevante: Embolia pulmonar, neumotórax, hemoptisis, asma, disnea inducida por esfuerzo. Trastornos gastrointestinales: Muy común / todos los grados: Diarrea, vómitos, náuseas, estomatitis, dolor abdominal. Común / todos los grados: Hemorragia gastrointestinal, estreńimiento, dolor abdominal superior, dispepsia, flatulencia, sequedad bucal. Poco común / grave o potencialmente fatal (grado 3-4) o considerado médicamente relevante: obstrucción intestinal, ascitis, enteritis, gastritis, disfagia, dolor abdominal inferior, esofagitis, reflujo gastroesofágico, colitis, sangre en heces. Trastornos hepatobiliares: Común / todos los grados: Hiperbilirrubinemia, anormalidad en pruebas de función hepática. Poco común / grave o potencialmente fatal (grado 3-4) o considerado médicamente relevante: Ictericia. Trastornos de la piel y tejido subcutáneo: Muy común / todos los grados: Síndrome de eritrodisestesia palmar-plantar. Común / todos los grados: rush, alopecia, eritema, piel seca, prurito, hiperpigmentación de la piel, urticaria macular, descamación de la piel, dermatitis, alteraciones de la pigmentación, alteraciones en las uńas. Poco común / grave o potencialmente fatal (grado 3-4) o considerado médicamente relevante: ampollar, úlceras de la piel, urticaria, reacción de fotosensibilidad, eritema palmar, hinchazón en rostro, púrpura, síndrome de hipersensibilización a la radiación. Trastornos músculo-esqueléticos y del tejido conectivo: Común / todos los grados: Dolor en extremidades, dolor de espalda, artralgia. Poco común / grave o potencialmente fatal (grado 3-4) o considerado médicamente relevante: inflamación en articulaciones, dolor de huesos, dolor facial, rigidez músculo-esquelética, debilidad muscular. Trastornos renales y urinarios: Poco común / grave o potencialmente fatal (grado 3-4) o considerado médicamente relevante: Hidronefrosis, incontinencia urinaria, hematuria, nocturia, incremento en creatinina sérica. Trastornos del sistema reproductivo y mamarios: Poco común / grave o potencialmente fatal (grado 3-4) o considerado médicamente relevante: hemorragia vaginal. Trastornos generales y condiciones del lugar de administración: Muy común / todos los grados: Fatiga, astenia. Común / todos los grados: Pirexia, edema periférico, malestar, dolor en el pecho, letargia. Poco común / grave o potencialmente fatal (grado 3-4) o considerado médicamente relevante: edema, escalofríos, enfermedad similar a la influenza, rigidez, incremento en temperatura corporal. Injurias, intoxicación y complicaciones de procedimiento: Poco común/ grave potencialmente fatal ( grado 3-4) o considerado medicamente relevante: ampollas, sobredosis. Capecitabina en terapia de combinación: La lista 2 a continuación muestra los ADR asociados con el uso de capecitabina en combinación con diferentes regímenes de quimioterapia en múltiples indicaciones en base a los datos de seguridad en mas de 3000 pacientes. Los ADR se ańaden al grupo de frecuencia adecuado (Muy común o Común) de acuerdo con la mayor incidencia observada en cualquiera de los ensayos clínicos y solo cuando se observan adicionalmente a los observados con capecitabina como monoterapia u observados en una mayor frecuencia en comparación con los observados con capecitabina como monoterapia (ver lista 1). Los ADR poco comunes reportados para capecitabina en terapia de combinación son consistentes con los ADR reportados para capecitabina como monoterapia o reportados para monoterapia con el producto farmacéutico de combinación (en la literatura y/o el resumen de características del producto respectivas). Algunos ADR son reacciones comúnmente observadas con el producto farmacéutico de combinación (como neuropatía sensorial periférica con docetaxel u oxaliplatino, hipertensión observada con bevacizumab); sin embargo, no puede excluirse una exacerbación con la terapia con capecitabina. Lista 2: Resumen de ADR reportados en pacientes tratados con capecitabina en terapia de combinación adicionalmente a los observados con monoterapia con capecitabina u observados en un grupo de mayor frecuencia en comparación con capecitabina como monoterapia. Infecciones e infestaciones: Común / todos los grados: Herpes zoster, infección del tracto urinario, candidiasis oral, infección del tracto respiratorio superior, rinitis, influenza, + infección, herpes oral. Trastornos de la sangre y sistema linfático: Muy común / todos los grados: +Neutropenia, +leucopenia, +anemia, +fiebre neutropénica, trombocitopenia. Común / todos los grados: Depresión de la médula ósea, +neutropenia febril. Trastornos del sistema inmunológico: Común / todos los grados: Hipersensibilidad. Trastornos del metabolismo y nutrición:Muy común / todos los grados: disminución del apetito. Común / todos los grados: hipokalemia, hiponatremia, hipomagnesemia, hipocalcemia, hiperglicemia. Trastornos psiquiátricos: Común / todos los grados: desórdenes de sueńo, ansiedad. Trastornos del sistema nervioso: Muy Común / todos los grados: Parestesia, disestesia, neuropatía periférica, neuropatía sensorial periférica, disgeusia, dolor de cabeza. Común / todos los grados: Neurotoxicidad, temblor, neuralgia, reacción de hipersensibilidad, hipostesia. Trastornos oculares: Muy común / todos los grados: Incremento en lagrimeo. Común / todos los grados: alteraciones visuales, ojo seco, dolor ocular, deterioro de la visión, visión borrosa. Trastornos del oído y laberinto: Común / todos los grados: Tinitus, hipoacusia. Trastornos cardíacos: Común / todos los grados: fibrilación auricular, isquemia/infarto cardíaco. Trastornos vasculares: Muy común / todos los grados: edema en miembros inferiores, hipertensión, +embolia y trombosis. Común / todos los grados: Rubor, hipotensión, crisis hipertensiva, bochornos, flebitis. Trastornos respiratorios, torácicos y del mediastino: Muy común / todos los grados: dolor de garganta, disestesia faríngea. Común / todos los grados: Hipo, dolor faringo-laringeo, disfonía. Trastornos gastrointestinales: Muy común / todos los grados: Estreńimiento, dispepsia. Común / todos los grados: hemorragia gastrointestinal superior, ulceración bucal, gastritis, distensión abdominal, enfermedad de reflujo gastroesofágico, dolor bucal, disfagia, hemorragia rectal, dolor abdominal inferior, disestesia oral, parestesia oral, hipostesia oral, malestar abdominal. Trastornos hepatobiliares: Común / todos los grados: Función hepática anormal. Trastornos de la piel y tejido subcutáneo: Muy común / todos los grados: Alopecia, alteraciones en las uńas. Común / todos los grados: Hiperhidrosis, rush eritematosa, urticaria, sudoración nocturnos. Trastornos músculo-esqueléticos y del tejido conectivo: Muy común / todos los grados: Mialgia, artralgia, dolor en extremidades. Común / todos los grados: Dolor de mandíbula, espasmos musculares, trismos, debilidad muscular. Trastornos renales y urinarios: Común / todos los grados: Hematuria, proteinuria, disminución en depuración de creatinina renal, disuria. Trastornos generales y condiciones del lugar de administración: Muy común / todos los grados: Pirexia, debilidad, +letargo, intolerancia a la temperatura. Común / todos los grados: Inflamación a las mucosas, dolor en extremidades, dolor, escalofríos, enfermedad similar a influenza, +fiebre, reacción relacionada con la infusión, reacción en el lugar de la inyección, dolor en el lugar de la inyección. Lesiones, intoxicación y complicaciones del procedimiento: Común / todos los grados: Contusión + para cada término el conteo de frecuencia se basó en los ADR de todos los grados. Para términos marcados con "+", la frecuencia se basó en ADR de grado 3 y 4. Las ADR se ańadieron de acuerdo con la incidencia más alta observada en cualquiera de los ensayos de combinación. Experiencia post-venta: Se han identificado las siguientes reacciones adversas graves adicionales durante la exposición post-venta: Lista 3: Resumen de eventos reportados con capecitabina en el esquema post-venta. Trastornos oculares: Raro: Estenosis del ducto lacrimal, alteraciones de la córnea, queratitis, queratitis puntuada. Trastornos cardíacos: Raro: Fibrilación ventricular, prolongación QT, taquicardia ventricular poliforme, vasoespasmo. Trastornos hepatobiliares: Raro: falla hepática, hepatitis colestática. Trastornos de la piel y subcutáneos: Raro: eritema lupus cutáneo. Muy raro: Reacciones graves a la piel como el síndrome de Stevens-Johnson y necrólisis epidérmica tóxica (ver sección Advertencias especiales y precauciones especiales para el uso). Trastornos renales y urinarios: Raro: Falla renal aguda secundaria a la deshidratación. Descripción de reacciones adversas seleccionadas: Síndrome mano-pie (HFS) (ver sección Advertencias especiales y precauciones especiales para el uso): Para la dosis de capecitabina de 1250 mg/m2 2 veces al día en los días 1 a 14 cada 3 semanas, se observó una frecuencia de 53% a 60% de HFS para todos los grados en ensayos con capecitabina monoterapia (incluyendo estudios de terapia coadyuvante en cáncer de colon, tratamiento de cáncer colorrectal metastásico y tratamiento de cáncer mamario) y una frecuencia de 63% en el grupo capecitabina/docetaxel para el tratamiento de cáncer mamario metastásico. Para la dosis de capecitabina de 1000 mg/m2 2 veces al día en los días 1 a 14 cada 3 semanas, se observó una frecuencia de 22% a 30% de HFS para todos los grados en la terapia combinada con capecitabina. Un metaanálisis de 14 ensayos clínicos con datos de más de 4700 pacientes tratados con capecitabina monoterapia o bien capecitabina en combinación con diferentes regímenes de quimioterapia en indicaciones múltiples (cáncer de colon, colorrectal, gástrico y de mama) mostró que el HFS (todos los grados) ocurrió en 2066 pacientes (43%) después de una mediana de tiempo de 239 [IC 95% 201, 288] días después de iniciar el tratamiento con capecitabina. En todos los estudios combinados, las siguientes covariables fueron estadística y significativamente asociadas a un aumento en el riesgo de desarrollar HFS: dosis inicial creciente de capecitabina (g), dosis acumulada decreciente de capecitabina (0.1*kg), intensidad de dosis relativa creciente en las primeras 6 semanas, duración creciente del tratamiento en estudio (semanas), edad creciente (en incrementos de 10 ańos), sexo femenino y un buen rendimiento ECOG a nivel basal (0 versus ł 1). Diarrea (ver sección Advertencias especiales y precauciones especiales para el uso): Capecitabina puede inducir la ocurrencia de diarrea, la cual se ha observado hasta en el 50% de los pacientes. Los resultados de un metaanálisis de 14 ensayos clínicos con datos de más de 4700 pacientes tratados con capecitabina mostraron que en todos los estudios combinados, las siguientes covariables fueron estadística y significativamente asociadas a un mayor riesgo de desarrollar diarrea: dosis inicial creciente de capecitabina (g), duración creciente del tratamiento en estudio (semanas), edad creciente (en incrementos de 10 ańos) y sexo femenino. Las siguientes covariables fueron estadística y significativamente asociadas a un menor riesgo de desarrollar diarrea: dosis acumulada creciente de capecitabina (0.1*kg) e intensidad de dosis relativa creciente en las primeras 6 semanas. Cardiotoxicidad (ver sección Advertencias especiales y precauciones especiales para el uso): Además de las ADRs descritas en las Listas 1 y 2, las siguientes ADRs con una incidencia menor al 0.1% se asociaron al uso de capecitabina monoterapia, en base a un análisis combinado de datos de seguridad clínica provenientes de 7 ensayos clínicos que incluyeron 949 pacientes (2 ensayos clínicos fase III y 5 fase II en cáncer colorrectal metastásico y cáncer mamario metastásico): cardiomiopatía, insuficiencia cardíaca, muerte súbita y extrasístole ventricular. Encefalopatía: Además de las ADRs descritas en las Listas 1 y 2, y en base al análisis combinado anterior de datos de seguridad clínica provenientes de 7 ensayos clínicos, la encefalopatía también se asoció al uso de capecitabina monoterapia, con una incidencia menor al 0.1%. Poblaciones especiales: Pacientes ancianos (ver sección Posología): Un análisis de datos de seguridad en pacientes ł 60 ańos de edad tratados con capecitabina monoterapia y un análisis de pacientes tratados con capecitabina más docetaxel combinados mostraron un incremento en la incidencia de reacciones adversas grado 3 y 4 asociadas al tratamiento y reacciones adversas graves asociadas al tratamiento en comparación con pacientes <60 ańos de edad. Los pacientes ł 60 ańos de edad tratados con capecitabina más docetaxel tuvieron además discontinuaciones tempranas del tratamiento debido a reacciones adversas en comparación con pacientes <60 ańos de edad. Los resultados de un metaanálisis de 14 ensayos clínicos con datos de más de 4700 pacientes tratados con capecitabina mostraron que en todos los estudios combinados, el incremento de la edad (en aumentos de 10 ańos) se asoció estadística y significativamente a un mayor riesgo de desarrollar HFS y diarrea, y a un menor riesgo de desarrollar neutropenia. Sexo: Los resultados de un metaanálisis de 14 ensayos clínicos con datos de más de 4700 pacientes tratados con capecitabina mostraron que en todos los estudios combinados, el sexo femenino se asoció estadística y significativamente a un mayor riesgo de desarrollar HFS y diarrea, y a un menor riesgo de desarrollar neutropenia. Pacientes con dańo renal (ver sección Posología, Advertencias especiales y precauciones especiales para el uso y Propiedades farmacocinéticas): Un análisis de datos de seguridad en pacientes tratados con capecitabina monoterapia (cáncer colorrectal) con dańo renal basal mostró un aumento en la incidencia de reacciones adversas grado 3 y 4 asociadas al tratamiento en comparación con pacientes con una función renal normal (36% en pacientes sin dańo renal n=268, vs. 41% en dańo leve n=257 and 54% n=59, en dańo moderado respectivamente) (ver sección Propiedades farmacocinéticas). Los pacientes con función renal con dańo moderado muestran un mayor porcentaje de reducción de dosis (44%) vs. 33% y 32% en pacientes con dańo renal leve o ausente y un incremento en las discontinuaciones tempranas del tratamiento (21% de discontinuaciones durante los primeros 2 ciclos) vs. 5% y 8% en pacientes con dańo renal leve o ausente.
|
|
Contraindicaciones:
Historia de reacciones graves e inesperadas a terapia con fluoropirimidinas. Hipersensibilidad a capecitabina o alguno de los excipientes detallados en la sección Lista de excipientes, o fluorouracilo. En pacientes con deficiencia conocida de dihidropirimidina deshidrogenasa (DPD). Durante el embarazo y lactancia. En pacientes con leucopenia, neutropenia, o trombocitopenia severa. En pacientes con dańo hepático severo. En pacientes con dańo renal severo (depuración de creatinina menor a 30 ml/min). Tratamiento con sorivudina o sus análogos químicamente relacionados, como ser brivudina (ver sección Interacciones medicamentosas). Si existen contraindicaciones para alguno de los agentes en el régimen combinado, ese agente no debe ser utilizado.
|
|
Advertencias:
Advertencias especiales y precauciones de uso: Las toxicidades limitadoras de dosis incluyen diarrea, dolor abdominal, náuseas, estomatitis y síndrome mano-pie (reacción cutánea mano-pie, eritrodisestesia palmar-plantar). La mayoría de las reacciones adversas son reversibles y no requieren la discontinuación permanente de la terapia, aunque podría ser necesario suspender o disminuir las dosis. Diarrea: Los pacientes con diarrea severa deben ser cuidadosamente monitoreados y recibir reemplazo de líquidos y electrolitos en caso de deshidratación. Se podrían emplear tratamientos antidiarreicos estándar (como la loperamida). La diarrea grado 2 NCIC CTC se define como un aumento de 4 a 6 deposiciones/día o deposiciones nocturnas, la diarrea grado 3 como un incremento de 7 a 9 deposiciones/día o incontinencia y mala absorción. La diarrea grado 4 es un incremento ł 10 deposiciones/día o diarrea con abundante sangre o bien la necesidad de soporte parenteral. Se debe aplicar reducción de dosis según necesidad (ver sección Posología). Deshidratación: La deshidratación debe ser prevenida o corregida al momento de su aparición. Pacientes con anorexia, astenia, náusea, vómitos o diarrea podrían deshidratarse con rapidez. Si ocurre una deshidratación grado 2 (o mayor), el tratamiento con capecitabina debe ser interrumpido de inmediato y corregir la deshidratación. El tratamiento no debe ser reiniciado hasta que el paciente se vuelva a hidratar y las causas hayan sido corregidas o controladas. Se deben aplicar modificaciones de las dosis para eventos adversos precipitantes según necesidad (ver sección Posología). Síndrome mano-pie (también conocido como reacción cutánea mano-pie o eritrodisestesia palmar -plantar o eritema acral inducido por quimioterapia): El síndrome mano-pie grado 1 se define como adormecimiento, disestesia/parestesia, hormigueo, hinchazón indolora o eritema de manos y/o pies y/o malestar que no altera las actividades normales del paciente. El síndrome mano-pie grado 2 es eritema doloroso e hinchazón de las manos y/o los pies y/o malestar que afecta las actividades de la vida diaria del paciente. El síndrome mano-pie grado 3 consiste en descamación húmeda, ulceración, formación de ampollas y dolor intenso de las manos y/o los pies y/o malestar severo que le impide al paciente trabajar o realizar actividades en su vida diaria. Si ocurre un síndrome mano-pie grado 2 ó 3, la administración de capecitabina debe ser interrumpida hasta que el evento se resuelva o disminuya de intensidad hasta el grado 1. Después de un síndrome mano-pie grado 3, las dosis posteriores de capecitabina deben disminuir. Al utilizar la asociación de capecitabina y cisplatino, no se recomienda el uso de vitamina B6 (piridoxina) para tratamiento profiláctico sintomático o secundario del síndrome mano-pie, ya que los informes publicados indican que podría reducir la eficacia del cisplatino. Cardiotoxicidad: La cardiotoxicidad ha sido asociada a terapia con fluoropirimidina, incluyendo infarto de miocardio, angina, disrritmias, shock cardiogénico, muerte súbita y cambios electrocardiográficos (incluyendo casos muy raros de prolongación QT). Estas reacciones adversas podrían ser más comunes en pacientes con antecedentes de enfermedad coronaria. Las arritmias cardíacas (incluyendo fibrilación ventricular, torsión de punta y bradicardia), angina de pecho, infarto de miocardio, insuficiencia cardiaca y cardiomiopatía han sido informados en pacientes que reciben capecitabina. Se debe tener precaución en pacientes con antecedentes de enfermedad cardíaca significativa, arritmias y angina de pecho (ver sección Efectos colaterales). Hipo o hipercalcemia: Se ha informado hipo o hipercalcemia durante el tratamiento con capecitabina. Se debe tener precaución en pacientes con hipo o hipercalcemia preexistente (ver sección Efectos colaterales). Enfermedad del sistema nervioso periférico o central: Se debe tener precaución en pacientes con enfermedad del sistema nervioso periférico o central, por ejemplo, metástasis cerebral o neuropatía (ver sección Efectos colaterales). Diabetes mellitus o alteración de electrolitos: Se debe tener precaución en pacientes con diabetes mellitus o alteración de electrolitos, ya que podrían verse agravados durante el tratamiento con capecitabina. Anticoagulantes derivados de cumarina: En un estudio de interacción farmacológica con administración de warfarina en dosis única, hubo un aumento significativo en el AUC medio (+57%) de S-warfarina. Estos resultados indican una interacción debida probablemente a una inhibición por capecitabina sobre el sistema de la isoenzima 2C9 del citocromo P450. Los pacientes que reciben en forma simultánea capecitabina y terapia anticoagulante oral derivada de cumarina deben recibir monitoreo cercano de su respuesta anticoagulante (INR o tiempo de protrombina) y ajustar la dosis de anticoagulante según corresponda (ver sección Interacciones medicamentosas). Dańo hepático: En ausencia de datos de eficacia y seguridad en pacientes con dańo hepático, el uso de capecitabina debe ser cuidadosamente monitoreado en pacientes con disfunción hepática leve a moderada, independiente de la presencia o ausencia de metástasis hepática. La administración de capecitabina debe ser interrumpida si ocurren elevaciones de bilirrubina asociadas al tratamiento >2.5 x ULN o elevaciones asociadas al tratamiento de aminotransferasas hepáticas (ALT, AST) de >2.5 x ULN. El tratamiento con capecitabina monoterapia puede ser retomado cuando la bilirrubina disminuye hasta Ł 3.0 x ULN o bien las aminotransferasas hepáticas disminuyen a Ł 2.5 x ULN. Dańo renal: La incidencia de reacciones adversas grado 3 ó 4 en pacientes con dańo renal moderado (depuración de creatinina 30-50 ml/min) aumenta en comparación con la población en general (ver sección Posología y Contraindicaciones). Deficiencia de DPD: Toxicidad rara, inesperada y grave (como estomatitis, diarrea, neutropenia y neurotoxicidad) asociada con 5-FU se ha atribuido a una deficiencia en la actividad de la DPD. No puede excluirse por tanto una relación entre los menores niveles de DPD y los mayores efectos tóxicos y potencialmente fatales del 5-FU. Los pacientes con deficiencia de DPD no deben ser tratados con capecitabina (ver sección Contraindicaciones). En pacientes con deficiencia de DPD no reconocida tratados con capecitabina, pueden ocurrir toxicidades que amenacen la vida como la sobredosis aguda (ver sección Sobredosificación). En caso de toxicidad aguda de grado 2-4, el tratamiendo debe ser discontinuado inmediatamente hasta que se resuelva la toxicidad observada. Debe considerarse la discontinuación permanente en base a la evaluación clínica de la aparición, duración y severidad de las toxicidades observadas. Complicaciones oftalmológicas: Los pacientes deben recibir cuidadoso monitoreo con respecto a complicaciones oftalmológicas tales como keratitis y desórdenes de la córnea, especialmente si tienen un historia anterior de desórdenes oculares. El tratamiento de desórdenes oculares debe ser iniciado en cuanto sea clínicamente adecuado. Reacciones cutáneas graves: la capecitabina puede inducir la aparición de reacciones cutáneas tales como el síndrome de Stevens-Johnson y necrolisis epidérmica tóxica. La capecitabina debe ser permanentemente discontinuada en pacientes que experimentan reacción cutánea grave durante el tratamiento. Dado que este medicamento contiene lactosa monohidrato como excipiente, los pacientes con problemas hereditarios raros de intolerancia a la galactosa, deficiencia de lactasa Lapp o mala absorción de glucosa-galactosa no deben usar este medicamento.
|
|
Precauciones:
Fertilidad, embarazo y lactancia: Mujeres fértiles: Se debe recomendar a las mujeres fértiles evitar el embarazo mientras reciben tratamiento con capecitabina. Si la paciente queda embarazada mientras recibe capecitabina, se debe explicar el riesgo potencial para el feto. Embarazo: No existen estudios en mujeres embarazadas usando capecitabina. Sin embargo, se debe suponer que capecitabina podría causar dańo fetal si se administra a mujeres embarazadas. En estudios de toxicidad reproductiva en animales, la administración de capecitabina causó letalidad embrionaria y teratogenicidad. Estos resultados son efectos esperados de los derivados de fluoropirimidinas. Capecitabina está contraindicada durante el embarazo. Lactancia: No se sabe si capecitabina es excretada en la leche materna. En ratones en período de lactancia se encontraron cantidades considerables de capecitabina y sus metabolitos en la leche. La lactancia debe ser discontinuada mientras se recibe tratamiento con capecitabina. Fertilidad: No existen datos sobre la capecitabina y su impacto en la fertilidad. Los estudios pivotales incluyeron mujeres y hombres con potencial para concebir solo si estos aceptaban usar un método anticonceptivo aceptable para evitar el embarazo durante el tiempo de duración del estudio y por un periodo razonable de tiempo luego de éste. En etudios animales se observaron efectos de los estudios en la fertilidad (ver sección Datos de seguridad preclínica). Efectos sobre la capacidad para conducir y usar maquinarias: Categor tiene una influencia menor o moderada sobre la capacidad para conducir y utilizar maquinaria. Capecitabina podría causar mareo, fatiga y náuseas.
|
|
Interacciones Medicamentosas:
Interacción con otros medicamentos y otras formas de interacción: Los estudios de interacción sólo han sido realizados en adultos. Interacción con otros medicamentos: Sustratos del citocromo P-450 2C9: Además de la warfarina, no se han realizado estudios de interacción fármaco-fármaco formales entre capecitabina y otros sustratos CYP2C9. Debe tener especial cuidado cuando la capecitabina es coadministrada con sustratos de 2C9 (como fenitoína). Revise también la interacción con anticoagulantes derivados de la cumarina a continuación y en la sección Advertencias especiales y precauciones especiales para el uso. Anticoagulantes derivados de la cumarina: parámetros de coagulación alterados y/o hemorragia han sido informados en pacientes que usan capecitabina en forma simultánea con anticoagulantes derivados de la cumarina como warfarina y fenprocumon. Estas reacciones ocurren dentro de varios días y hasta varios meses después de iniciada la terapia con capecitabina y, en unos pocos casos, dentro de 1 mes después de suspender la capecitabina. En un estudio clínico de interacción farmacocinética, después de una dosis única de 20 mg de warfarina, el tratamiento con capecitabina aumentó el AUC de S-warfarina en un 57%, con un 91% de incremento en el valor INR. Dado que el metabolismo de R-warfarina no fue afectado, estos resultados indican que capecitabina regula a la baja la isoenzima 2C9, pero no tiene efecto en las isoenzimas 1A2 y 3A4. Los pacientes que toman anticoagulantes derivados de la cumarina en forma simultánea con capecitabina deben ser monitoreados regularmente para detectar alteraciones en sus parámetros de coagulación (PT o INR) y ajustar la dosis de anticoagulante según corresponda. Fenitoína: se han informado mayores concentraciones plasmáticas de fenitoína que llevan a síntomas de intoxicación por fenitoína en casos individuales durante el uso concomitante de capecitabina con fenitoína. Los pacientes que toman fenitoína en forma simultánea con capecitabina deben tener un monitoreo regular para detectar concentraciones plasmáticas de fenitoína aumentadas. Acido folínico: un estudio de asociaciones con capecitabina y ácido folínico indicó que el ácido folínico no tiene efecto mayor en la farmacocinética de capecitabina y sus metabolitos. Sin embargo, el ácido folínico tiene efecto sobre las farmacodinámica de capecitabina y su toxicidad puede ser aumentada por el ácido folínico: la dosis tolerada máxima (MTD) de capecitabina sola utilizando el régimen intermitente es de 3000 mg/m2 al día, mientras que es solamente de 2000 mg/m2 al día al combinar capecitabina con ácido folínico (30 mg vía oral 2 veces al día). Sorivudina y análogos: se ha descrito una interacción farmacológica clínicamente significativa entre sorivudina y 5-FU, proveniente de la inhibición de la dihidropirimidina deshidrogenasa por sorivudina. Esta interacción, que lleva a una mayor toxicidad de la fluoropirimidina, es potencialmente fatal. En consecuencia, capecitabina no debe ser administrada en forma simultánea con sorivudina o sus análogos químicamente relacionados como la brivudina (ver sección Contraindicaciones). Debe existir al menos un período de espera de 4 semanas entre el término del tratamiento con sorivudina o sus análogos químicamente relacionados como la brivudina y el inicio de la terapia con capecitabina. Antiácidos: se investigó el efecto de un antiácido que contiene hidróxido de aluminio e hidróxido de magnesio sobre la farmacocinética de la capecitabina. Hubo un pequeńo aumento en las concentraciones plasmáticas de capecitabina y un metabolito (5'-DFCR); no hubo efecto sobre los 3 metabolitos principales (5'-DFUR, 5-FU y FBAL). Alopurinol: se han observado interacciones con alopurinol para 5-FU, con una posible disminución de la eficacia del 5-FU. El uso simultáneo de alopurinol con capecitabina debe ser evitado. Interacción con el citocromo P450: Para interacciones potenciales con las isoenzimas 1A2, 2C9 y 3A4, ver interacciones con anticoagulantes derivados de la cumarina. Interferón alfa: el MTD de capecitabina fue de 2000 mg/m2 al día al combinarse con el interferón alfa-2a (3 MIU/m2 al día), comparado con 3000 mg/m2 al día al utilizar capecitabina sola. Radioterapia: el MTD de capecitabina sola usando el régimen intermitente es de 3000 mg/m2 al día, mientras que, al combinarse con radioterapia para cáncer rectal, el MTD de capecitabina es de 2000 mg/m2 al día usando un esquema continuo o la administración diaria de lunes a viernes durante un ciclo de radioterapia de 6 semanas. Oxaliplatino: no hubo diferencias clínicamente significativas en la exposición a capecitabina o sus metabolitos, platino libre o platino total al administrar capecitabina asociada a oxaliplatino o en combinación con oxaliplatino y bevacizumab. Bevacizumab: no hubo un efecto clínicamente significativo de bevacizumab sobre los parámetros farmacocinéticos de capecitabina o sus metabolitos en presencia de oxaliplatino. Interacciones alimentarias: En todos los ensayos clínicos, se indicó a los pacientes administrar capecitabina dentro de los 30 minutos posteriores a una comida. Dado que los actuales datos de eficacia y seguridad se basan en la administración con alimentos, se recomienda administrar capecitabina con alimentos. De esta forma disminuye la velocidad de absorción de capecitabina (ver sección Propiedades farmacocinéticas).
|
|
Sobredosificación:
Las manifestaciones de sobredosis aguda incluyen náusea, vómitos, diarrea, mucositis, irritación y hemorragia gastrointestinal y depresión de la médula ósea. El manejo médico de la sobredosis debe incluir intervenciones médicas terapéuticas y de soporte personalizadas dirigidas a corregir las manifestaciones clínicas y prevenir sus posibles complicaciones.
|
|



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}