 |
Composición:
Cada cįpsula de 4 mg contiene: Pomalidomida. Excipientes: Manitol Almidón Pregelatinizado, Estearil Fumarato de Sodio, Gelatina, Dióxido Titanio Colorante FD&C Azul N° 2, Colorante FD&C Azul N° 1, Goma Laca, Dióxido de Titanio, Simeticona, Propilenglicol, Hidróxido de Amonio.
|
|
Acción Terapéutica:
Agente inmunomodulador.
|
|
Indicaciones:
Pomalyst® en combinación con dexametasona estį indicado en el tratamiento de los pacientes adultos con mieloma mśltiple resistente al tratamiento y recidivante que hayan recibido al menos 2 tratamientos previos, incluyendo lenalidomida y bortezomib, y que hayan experimentado una progresión de la enfermedad en el śltimo tratamiento.
|
|
Posologķa:
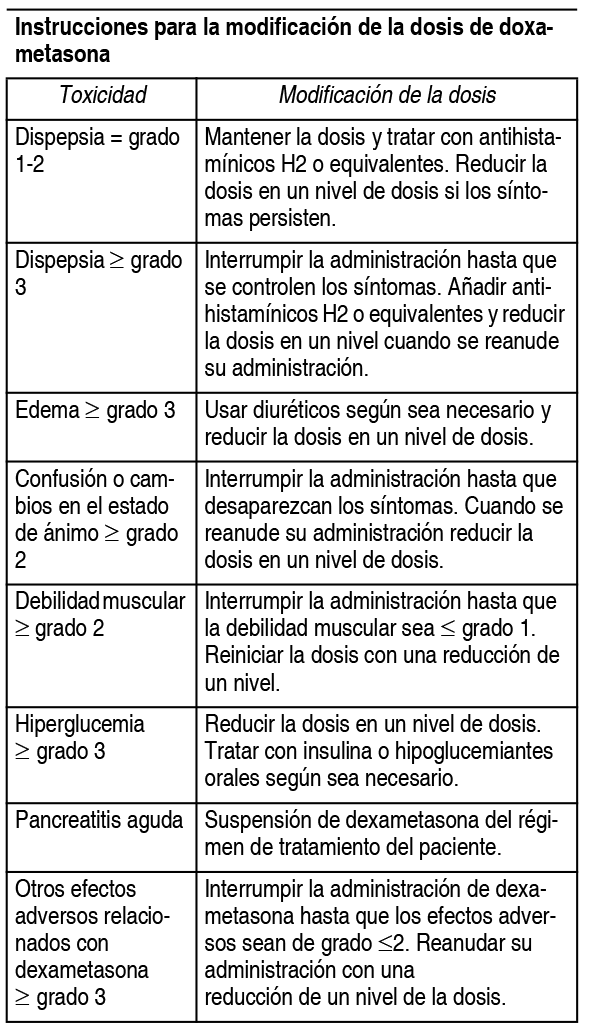
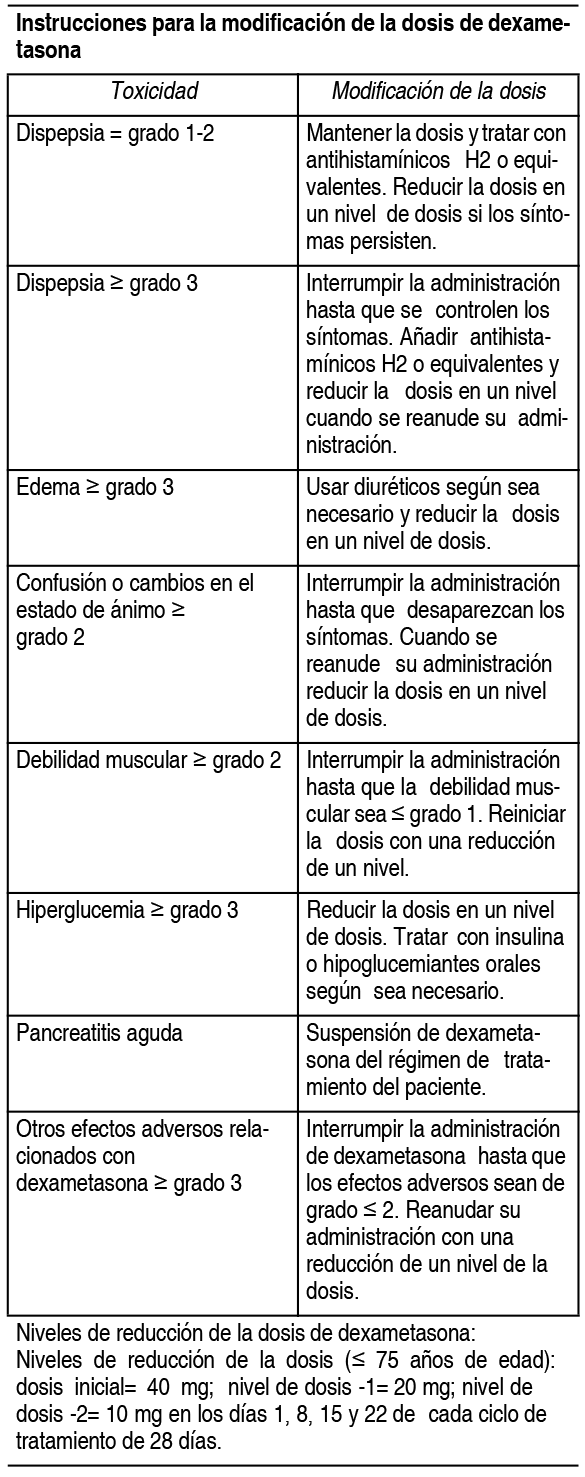
El tratamiento debe iniciarse y monitorizarse bajo la supervisión de médicos con experiencia en el tratamiento del mieloma mśltiple. Posologķa: La dosis inicial recomendada es de 4 mg de Pomalyst 1 vez al dķa por vķa oral, en los dķas del 1 al 21 de ciclos repetidos de 28 dķas. La dosis recomendada de dexametasona es de 40 mg por vķa oral 1 vez al dķa, en los dķas 1, 8, 15 y 22 de cada ciclo de tratamiento de 28 dķas. La posologķa se mantiene o modifica en función de los resultados clķnicos y de laboratorio. El tratamiento debe suspenderse si existe evidencia de progresión de la enfermedad. Modificación o interrupción de la dosis de pomalidomida: Las instrucciones para la interrupción y reducción de la dosis de pomalidomida relacionada con reacciones adversas hematológicas se indican en la siguiente tabla:
Ver Tabla
Para iniciar un nuevo ciclo de pomalidomida, el recuento de neutrófilos debe ser ³ 1 x 109/l y el recuento de plaquetas debe ser ³ 50 x 109/l. En caso de neutropenia, el médico debe considerar el uso de factores de crecimiento. En el caso de otras reacciones adversas de grado 3 ó 4 relacionadas con pomalidomida, el médico debe considerar interrumpir el tratamiento y reanudarlo con un 1 mg menos que la dosis previa una vez que se haya disminuido la reacción adversa a un grado inferior o igual a 2. Si la reacción adversa ocurre tras disminuciones de la dosis a 1 mg, entonces debe suspenderse el tratamiento con este medicamento.
Ver Tabla
Se debe considerar la interrupción o suspensión de pomalidomida en caso de exantema de grado 2-3. Se debe suspender el tratamiento con pomalidomida en caso de angioedema, exantema de grado 4 y exantema ampolloso o exfoliativo, y no se debe reanudar una vez suspendido por estas reacciones. Si se administran inhibidores potentes del CYP1A2 (p. ej., ciprofloxacino, enoxacino y fluvoxamina) de forma concomitante con pomalidomida, se debe reducir la dosis de pomalidomida en un 50%.
Ver Tabla
Niveles de reducción de la dosis de dexametasona: Niveles de reducción de la dosis ( £ 75 ańos de edad): dosis inicial= 40 mg; nivel de dosis -1= 20 mg; nivel de dosis -2= 10 mg en los dķas 1, 8, 15 y 22 de cada ciclo de tratamiento de 28 dķas. Niveles de reducción de la dosis (>75 ańos de edad): dosis inicial= 20 mg; nivel de dosis -1= 12 mg; nivel de dosis -2= 8 mg en los dķas 1, 8, 15 y 22 de cada ciclo de tratamiento de 28 dķas. Si la recuperación de las toxicidades tarda mįs de 14 dķas, la dosis de dexametasona se reducirį en un nivel de dosis. Poblaciones especiales: Población pediįtrica: No existe una recomendación de uso especķfica para Pomalyst en nińos de 0 a 17 ańos para la indicación del mieloma mśltiple. Pacientes de edad avanzada: No se requiere ningśn ajuste de dosis de pomalidomida. En pacientes de >75 ańos de edad, la dosis inicial de dexametasona es de 20 mg 1 vez al dķa en los dķas 1, 8, 15 y 22 de cada ciclo de tratamiento de 28 dķas. Insuficiencia renal: No se ha realizado ningśn estudio sobre el uso de pomalidomida en sujetos con insuficiencia renal. Los pacientes con insuficiencia renal moderada o grave (aclaramiento de creatinina <45 ml/min) se excluyeron de los estudios clķnicos. Los pacientes con insuficiencia renal deben ser monitorizados cuidadosamente por la aparición de reacciones adversas. Insuficiencia hepįtica: No se ha realizado ningśn estudio sobre el uso de pomalidomida en sujetos con insuficiencia hepįtica. Los pacientes con una concentración sérica de bilirrubina total >2.0 mg/dl se excluyeron de los estudios clķnicos. Los pacientes con insuficiencia hepįtica deben ser monitorizados cuidadosamente por la aparición de reacciones adversas. Forma de administración: Vķa oral. Pomalyst debe tomarse a la misma hora cada dķa. Las cįpsulas no deben abrirse, romperse ni masticarse. Este medicamento debe tomarse entero, preferiblemente con agua, con o sin alimentos. Si el paciente olvida tomar una dosis de Pomalyst 1 dķa, el paciente debe entonces tomar la próxima dosis al dķa siguiente a la hora habitual. Los pacientes no deben ajustar la dosis para compensar una dosis olvidada en dķas anteriores.
|
|
Efectos Colaterales:
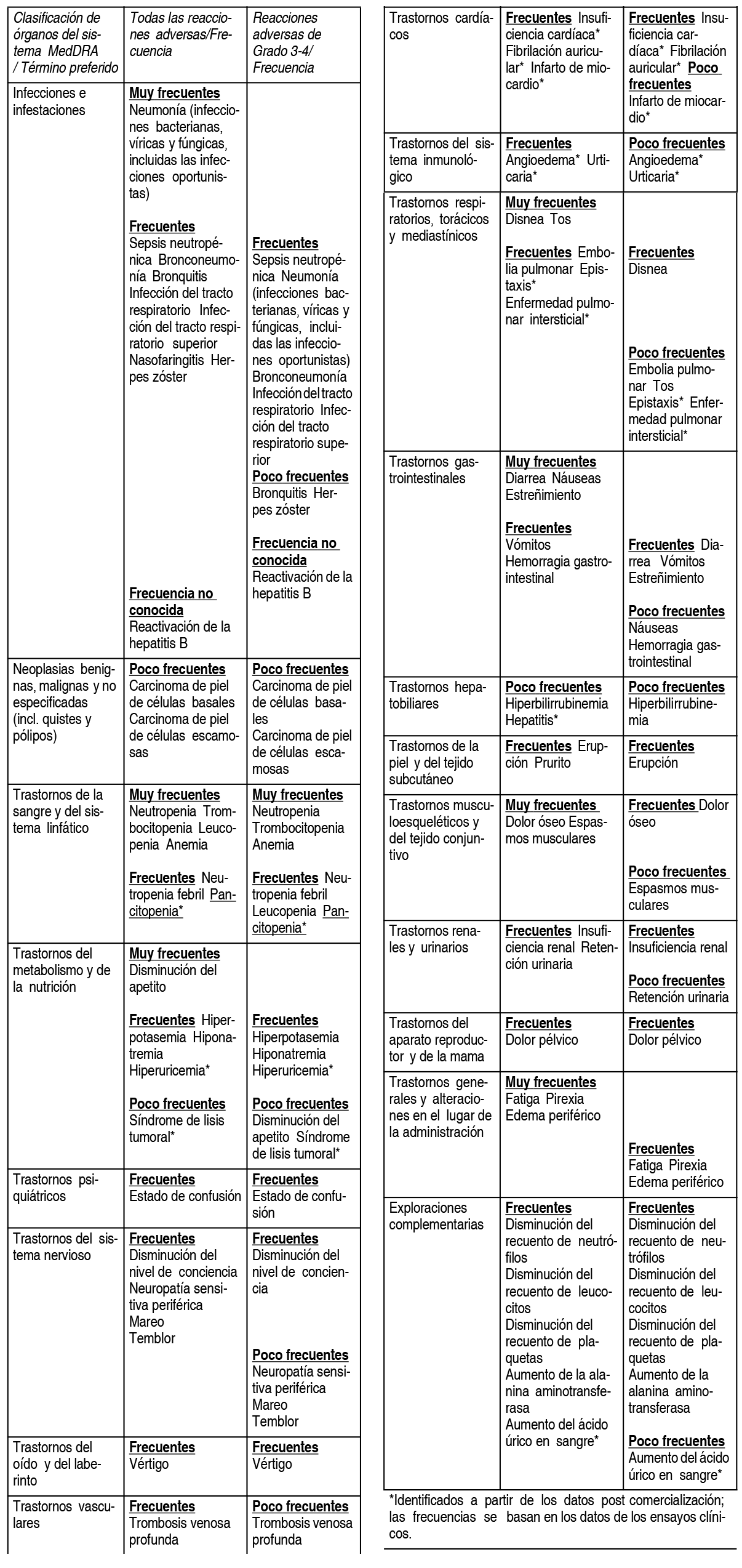
Resumen del perfil de seguridad: Las reacciones adversas notificadas con mayor frecuencia en los estudios clķnicos han sido los trastornos de la sangre y del sistema linfįtico, incluyendo anemia (45.7%), neutropenia (45.3%) y trombocitopenia (27%); trastornos generales y alteraciones en el lugar de la administración, incluyendo fatiga (28.3%), pirexia (21%) y edema periférico (13%); e infecciones e infestaciones incluyendo neumonķa (10.7%). Las reacciones adversas relacionadas con neuropatķa periférica fueron notificadas en el 123% de los pacientes y las reacciones adversas de embolismo o tromboembolismo venoso fueron notificadas en el 3.3% de los pacientes. Las reacciones adversas de grado 3 ó 4 mįs frecuentes estaban relacionadas con trastornos de la sangre y del sistema linfįtico, incluyendo neutropenia (41.7%), anemia (27%) y trombocitopenia (20.7%); infecciones e infestaciones, incluyendo neumonķa (9%); y trastornos generales y alteraciones en el lugar de la administración, incluyendo fatiga (4.7%), pirexia (3%) y edema periférico (1.3%). La reacción adversa grave notificada con mayor frecuencia fue la neumonķa (9.3%). Otras reacciones adversas graves notificadas incluyen neutropenia febril (4.0%), neutropenia (2.0%), trombocitopenia (1.7%) y reacciones adversas de TEV (1.7%). Se observó que las reacciones adversas tendķan a ocurrir con mayor frecuencia dentro de los primeros 2 ciclos de tratamiento con pomalidomida. Lista tabulada de reacciones adversas: En el estudio aleatorizado CC-4047-MM-003, un total de 302 pacientes con mieloma mśltiple en recaķda y refractario fueron tratados con 4 mg de pomalidomida administrada 1 vez al dķa durante 21 dķas en cada ciclo de 28 dķas, en combinación con una dosis baja semanal de dexametasona. Las reacciones adversas observadas en los pacientes tratados con pomalidomida y dexametasona se incluyen a continuación, segśn el sistema de clasificación por órganos y la frecuencia (SOC por sus siglas en inglés) para todas las reacciones adversas y para las reacciones adversas de Grado 3 ó 4. Las frecuencias de las reacciones adversas son las notificadas en el grupo de pomalidomida mįs dexametasona del estudio CC-4047-MM-003 (n=302) y de los datos poscomercialización. Las reacciones adversas se presentan en orden decreciente de gravedad dentro de cada intervalo de SOC (por sus siglas en inglés) y de frecuencia. Las frecuencias se definen, segśn las guķas actuales, como: muy frecuentes ( ³ 1/10); frecuentes ( ³ 1/100 a < 1/10) y poco frecuentes ( ³ 1/1.000 a <1/100).
Ver Tabla
Descripción de reacciones adversas seleccionadas: Teratogenicidad: Pomalidomida estį relacionada estructuralmente con talidomida. Talidomida es un principio activo con acción teratogénica conocida en humanos, que causa defectos de nacimiento graves que pueden poner en peligro la vida del nińo. Pomalidomida tiene un efecto teratogénico en ratas y conejos cuando se administra durante el perķodo de mayor organogénesis. Si se toma pomalidomida durante el embarazo, se espera un efecto teratogénico de pomalidomida en los seres humanos. Neutropenia y trombocitopenia: El 45.3% de los pacientes que recibieron pomalidomida mįs dosis bajas de dexametasona (Pom + LD-Dex) experimentó neutropenia, frente al 19.5% de los pacientes que recibieron dosis altas de dexametasona (HD-Dex). La neutropenia fue de grado 3 ó 4 en un 41.7% de los pacientes que recibieron Pom + LD-Dex frente al 14.8% de los que recibieron HD-Dex. En los pacientes tratados con Pom + LD-Dex, la neutropenia fue grave en una minorķa (2.0% de los pacientes), no tuvo como consecuencia la suspensión del tratamiento, y se asoció con la interrupción del tratamiento en un 21.0% de los pacientes y con una reducción de la dosis en un 7.7% de los pacientes. El 6.7% de los pacientes que recibieron Pom + LD-Dex experimentó neutropenia febril (NF), frente a ninguno de aquéllos que recibieron HD-Dex. Todos los casos fueron notificados como de grado 3 ó 4. La NF fue notificada como grave en un 4.0% de los pacientes. Asimismo, la NF se asoció con una interrupción de la dosis en un 3.7% de los pacientes, con una reducción de la dosis en un 1.3% de los pacientes y con ningśn caso de suspensión del tratamiento. El 27.0% de los pacientes que recibieron Pom + LD-Dex experimentó trombocitopenia frente a un 26.8% de los pacientes que recibieron HD-Dex. La trombocitopenia fue de grado 3 ó 4 en un 20.7% de los pacientes que recibieron Pom + LD-Dex frente a un 24.2% de los que recibieron HD-Dex. En los pacientes tratados con Pom + LD-Dex, la trombocitopenia fue grave en un 1.7% de los pacientes, conllevó una reducción de la dosis en un 6.3% de los pacientes, una interrupción de la dosis en un 8% de los pacientes y la suspensión del tratamiento en un 0.7% de los pacientes. Infección: La infección fue la toxicidad no hematológica mįs frecuente, con una incidencia del 55.0% en los pacientes que recibieron Pom + LD-Dex frente al 48.3% en los pacientes que recibieron HD-Dex. Aproximadamente la mitad de dichas infecciones fueron de grado 3 ó 4; un 24.0% en los pacientes tratados con Pom + LD-Dex y un 22.8% en los pacientes tratados con HD- Dex. Entre los pacientes tratados con Pom + LD-Dex las infecciones notificadas con mayor frecuencia fueron la neumonķa y la infección del tracto respiratorio superior (en un 10.7% y un 9.3% de los pacientes, respectivamente); con un 24.3% de las infecciones notificadas clasificadas como graves o mortales (grado 5) en el 2.7% de los pacientes tratados. En los pacientes tratados con Pom + LD-Dex las infecciones conllevaron la suspensión de la dosis en un 2.0% de los pacientes, la interrupción del tratamiento en un 14.3% de los pacientes y la reducción de la dosis en un 1.3% de los pacientes. Eventos tromboembólicos: El 3.3% de los pacientes que recibieron Pom + LD-Dex y un 2.0% de los pacientes que recibieron HD-Dex experimentaron un embolismo o tromboembolismo venoso. El 1.3% de los pacientes que recibieron Pom + LD-Dex experimentó reacciones de grado 3 ó 4 frente a ningśn caso entre los pacientes que recibieron HD-Dex. En los pacientes tratados con Pom + LD- Dex, el TEV se notificó como grave en el 1.7%, no se notificó ninguna reacción adversa mortal en los estudios clķnicos y el TEV no se asoció con suspensión de la dosis. La profilaxis con įcido acetilsalicķlico (y otros anticoagulantes en pacientes de alto riesgo) fue obligatoria para todos los pacientes participantes en los estudios clķnicos. Se recomienda terapia anticoagulante (a no ser que esté contraindicada). Neuropatķa periférica: Se excluyó de los estudios clķnicos a los pacientes con neuropatķa periférica en curso de grado ³ 2. El 12.3% de los pacientes que recibieron Pom + LD-Dex experimentaron una neuropatķa periférica, la mayorķa de grado 1 ó 2, frente a un 10.7% de los pacientes que recibieron HD-Dex. El 1.0% de los pacientes que recibieron Pom + LD-Dex experimentó reacciones de grado 3 ó 4 frente al 1.3% de los pacientes que recibieron HD-Dex. En los estudios clķnicos, ninguna de las neuropatķas periféricas se notificó como grave en los pacientes tratados con Pom + LD-Dex y la neuropatķa periférica condujo a la suspensión de la dosis en un 0.3% de los pacientes. La mediana del tiempo hasta la aparición de la neuropatķa fue de 2.1 semanas, con una variación de 0.1 a 48.3 semanas. La mediana del tiempo hasta la aparición fue inferior en los pacientes que recibieron HD-Dex comparado con los pacientes tratados con Pom+LD-Dex (1.3 semanas frente a 2.1 semanas). La mediana del tiempo hasta la desaparición de los sķntomas fue de 22.4 semanas en los pacientes que recibieron Pom+LD-Dex y de 13,6 semanas en los pacientes que recibieron HD-Dex. El lķmite inferior del intervalo de confianza del 95% fue de 5.3 semanas en los pacientes tratados con Pom+LD-Dex y de 2.0 semanas en los pacientes que recibieron HD-Dex. Hemorragia: Se han notificado trastornos hemorrįgicos con pomalidomida, especialmente en pacientes con factores de riesgo tales como medicamentos concomitantes que aumentan el riesgo de hemorragia. Los eventos hemorrįgicos incluyen epistaxis, hemorragia intracraneal y hemorragia gastrointestinal. Notificación de efectos adversos: Si experimenta cualquier tipo de efecto adverso, consulte a su médico, farmacéutico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este prospecto. También puede comunicarlos directamente. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar mįs información sobre la seguridad de este medicamento. Para mįs información, llame al laboratorio Tecnofarma al teléfono 225949201.
|
|
Contraindicaciones:
Embarazo. Mujeres con capacidad de gestación, a menos que se cumplan todas las condiciones del Programa de Prevención de Embarazo. Pacientes varones incapaces de seguir o cumplir las medidas anticonceptivas requeridas. Hipersensibilidad al principio activo o a alguno de los excipientes.
|
|
Advertencias:
Teratogenicidad: Pomalidomida no debe tomarse durante el embarazo ya que es esperable un efecto teratogénico. Pomalidomida estį relacionada estructuralmente con talidomida. Talidomida es un teratógeno conocido en humanos, que causa defectos congénitos de nacimiento graves que pueden poner en peligro la vida del nińo. Pomalidomida tiene un efecto teratogénico en ratas y conejos cuando se administra durante el perķodo de mayor organogénesis. Todas las pacientes deben cumplir las condiciones del Programa de Prevención de Embarazo a menos que exista evidencia fiable de que la paciente no tiene capacidad de gestación. Criterios para definir a las mujeres que no tienen capacidad de gestación: Se considera que una paciente o la pareja de un paciente varón no tiene capacidad de gestación si cumple por lo menos 1 de los siguientes criterios: Edad ³ 50 ańos y con amenorrea natural durante ³ 2 ańos*. Insuficiencia ovįrica prematura confirmada por un ginecólogo especialista. Salpingooforectomķa bilateral o histerectomķa previas. Genotipo XY, sķndrome de Turner, agenesia uterina. *La amenorrea que pueda aparecer después de un tratamiento oncológico o durante la lactancia no descarta la capacidad de gestación. Asesoramiento: En mujeres con capacidad de gestación, pomalidomida estį contraindicada a menos que la paciente cumpla todas las condiciones que se indican a continuación: Comprende el riesgo teratogénico esperado para el feto. Comprende la necesidad de utilizar dos (2) métodos anticonceptivos eficaces, sin interrupción, desde 4 semanas antes de iniciar el tratamiento, durante la duración completa del mismo y 4 semanas después de finalizarlo. Incluso si una mujer con capacidad de gestación tiene amenorrea, debe seguir todos los consejos sobre anticoncepción eficaz. Debe ser capaz de cumplir las medidas anticonceptivas eficaces. Estį informada y comprende las potenciales consecuencias del embarazo, y la necesidad de consultar rįpidamente a un especialista si hay riesgo de embarazo. Comprende la necesidad de comenzar a utilizar métodos anticonceptivos tan pronto como se le dispense pomalidomida y tras haber obtenido un resultado negativo en la prueba de embarazo. Comprende la necesidad de realizar pruebas de embarazo y acepta hacérselas cada 4 semanas, excepto en el caso de que se halla sometido previamente a una ligadura de trompas de eficacia confirmada. Confirma que comprende los peligros y las precauciones necesarias asociadas al uso de pomalidomida. El médico prescriptor debe comprobar que, en el caso de las mujeres con capacidad de gestación: La paciente cumple las condiciones del Programa de Prevención de Embarazo, incluida la confirmación de que tiene un nivel de comprensión adecuado. La paciente ha aceptado las condiciones mencionadas anteriormente. En el caso de pacientes varones que toman pomalidomida, los datos farmacocinéticos han demostrado que pomalidomida estį presente en el semen humano. Como medida de precaución, todos los pacientes varones que tomen pomalidomida deben cumplir los siguientes requisitos: Comprende el riesgo teratogénico esperado si tiene relaciones sexuales con una mujer embarazada o con una mujer con capacidad de gestación. Comprende la necesidad del uso de preservativos si tiene relaciones sexuales con una mujer embarazada o con una mujer con capacidad de gestación que no utiliza métodos anticonceptivos eficaces, durante el tratamiento y durante los 7 dķas posteriores a la interrupción de la dosis y/o el cese del tratamiento. Los varones vasectomizados deben utilizar preservativos si tienen relaciones sexuales con una mujer embarazada ya que la pomalidomida puede estar presente en el semen aun en ausencia de espermatozoides.Comprende que si su pareja se queda embarazada mientras él estį tomando pomalidomida o durante los 7 dķas posteriores a la suspensión del tratamiento con pomalidomida, debe informar inmediatamente a su médico y es recomendable derivar a su pareja a un médico especialista o con experiencia en teratologķa para su evaluación y asesoramiento. Anticoncepción: Las mujeres con capacidad de gestación deben usar dos (2) métodos anticonceptivos eficaces desde 4 semanas antes del tratamiento, durante el tratamiento y hasta 4 semanas después del tratamiento con pomalidomida, e incluso en el caso de interrupción de la administración, a menos que la paciente se comprometa a mantener una abstinencia sexual absoluta y continua, que serį confirmada mensualmente. Si la paciente no utiliza dos (2) métodos anticonceptivos eficaces, debe ser derivada a un profesional sanitario debidamente capacitado con objeto de que reciba asesoramiento para empezar a utilizar métodos anticonceptivos. Los siguientes métodos pueden considerarse ejemplos de métodos anticonceptivos adecuados: Dispositivo intrauterino (DIU). Sistema de liberación intrauterino de levonorgestrel. Sistemas "depot" de liberación de acetato de medroxiprogesterona. Ligadura de trompas. Relaciones sexuales sólo con varones vasectomizados: la eficacia de la vasectomķa debe confirmarse mediante 2 anįlisis de semen negativos. Inhibidores de la ovulación que contienen progestįgeno solo (p. ej. desogestrel). Debido al riesgo aumentado de tromboembolismo venoso (TEV) en pacientes con mieloma mśltiple que toman pomalidomida y dexametasona, no se recomienda el uso concomitante de anticonceptivos orales combinados. Si una paciente estį tomando anticonceptivos orales combinados, debe cambiar a uno de los métodos anticonceptivos eficaces enumerados anteriormente. El riesgo aumentado de tromboembolismo venoso se mantiene durante un perķodo de 4 a 6 semanas después de suspender el tratamiento con anticonceptivos orales combinados. La eficacia de los anticonceptivos esteroideos puede verse reducida durante el tratamiento concomitante con dexametasona. Los dispositivos intrauterinos y los sistemas de liberación intrauterinos de levonorgestrel se asocian con un mayor riesgo de infección en el momento de la colocación y con hemorragia vaginal irregular. En especial en las pacientes con neutropenia debe considerarse el uso profilįctico de antibióticos. La colocación de dispositivos intrauterinos de liberación de cobre no estį recomendada, debido al potencial riesgo de infección en el momento de su colocación y a la pérdida de sangre menstrual, que pueden suponer un peligro para las pacientes con neutropenia grave o trombocitopenia grave. Pruebas de embarazo: Las mujeres con capacidad de gestación deben efectuarse pruebas de embarazo con una sensibilidad mķnima de 50 mUI/ml bajo supervisión médica y conforme a la prįctica habitual, tal como se explica a continuación. Este requisito incluye a las mujeres con capacidad de gestación que practican una abstinencia sexual absoluta y continua. Idealmente, la prueba de embarazo, la prescripción y la dispensación deben realizarse el mismo dķa. Pomalidomida se debe dispensar a las mujeres con capacidad de gestación en un plazo de 7 dķas tras la prescripción. Antes de iniciar el tratamiento: Debe efectuarse una prueba de embarazo bajo supervisión médica durante la consulta, en el momento de prescribir pomalidomida o en los 3 dķas anteriores a la visita al médico prescriptor, siempre que la paciente haya estado usando dos (2) métodos anticonceptivos eficaces durante al menos 4 semanas. La prueba debe garantizar que la paciente no esté embarazada cuando inicie el tratamiento con pomalidomida. Seguimiento y finalización del tratamiento: Se debe repetir cada 4 semanas una prueba de embarazo bajo supervisión médica, y realizar otra 4 semanas después de la finalización del tratamiento, excepto en el caso de que la paciente se halla sometido a una ligadura de trompas de eficacia confirmada. Estas pruebas de embarazo deben efectuarse el mismo dķa de la consulta en que se prescriba el medicamento o en los 3 dķas anteriores a la visita al médico prescriptor. Varones: Pomalidomida estį presente en el semen humano durante el tratamiento. Como medida de precaución, y teniendo en cuenta las poblaciones especiales con un tiempo de eliminación potencialmente prolongado, como la insuficiencia renal, todos los pacientes varones que tomen pomalidomida, incluyendo a aquellos que se hayan sometido a una vasectomķa, deben usar preservativos durante todo el tratamiento, durante la interrupción de la administración y hasta 7 dķas después del final del tratamiento, si su pareja estį embarazada o tiene capacidad de gestación y no estį usando ningśn método anticonceptivo. Los pacientes varones no deben donar semen o esperma durante el tratamiento (perķodos de interrupción de la dosis incluidos) ni en el plazo de 7 dķas después de la suspensión del tratamiento con pomalidomida. Precauciones adicionales: Se debe indicar a los pacientes que no den nunca este medicamento a otra persona y que devuelvan las cįpsulas sin usar al final del tratamiento. Los pacientes no deben donar sangre, semen o esperma durante el tratamiento (perķodos de interrupción de la dosis incluidos) ni en el plazo de 7 dķas después de la suspensión del tratamiento con pomalidomida. Material educativo, restricciones de prescripción y dispensación: Con objeto de ayudar a los pacientes a evitar la exposición fetal a pomalidomida, el Titular de la Autorización de Comercialización distribuirį material educativo a los profesionales sanitarios, destinado a reforzar las advertencias acerca de la teratogenicidad esperada de pomalidomida y a proporcionar asesoramiento sobre anticoncepción antes de iniciar el tratamiento y sobre la necesidad de realizar pruebas de embarazo. El médico debe informar a la paciente acerca del riesgo teratogénico esperado y de las estrictas medidas de prevención de embarazo, especificadas en el Programa de Prevención de Embarazo asķ como proporcionar a la paciente un folleto informativo adecuado, una tarjeta de paciente y/o una herramienta equivalente conforme al sistema nacional implementado de tarjeta del paciente. Se ha implementado un sistema de distribución nacional controlado. El sistema de distribución controlada incluye el uso de una tarjeta del paciente y/o un herramienta equivalente para el control de la prescripción y/o dispensación, asķ como la recolección de datos detallados en relación con la indicación terapéutica, para monitorizar el uso en una indicación no autorizada dentro del territorio nacional. Idealmente, la prueba de embarazo, la prescripción y la dispensación deben realizarse el mismo dķa. La dispensación de pomalidomida en mujeres con capacidad de gestación debe hacerse dentro de los 7 dķas de prescripción y tras haber obtenido un resultado negativo en la prueba de embarazo supervisada por un médico. Las prescripciones a mujeres con capacidad de gestación deben tener una cantidad mįxima de una (1) caja/frasco y las prescripciones para el resto de pacientes pueden tener una cantidad mįxima de dos (2) cajas/frascos. Eventos hematológicos: La reacción adversa hematológica de Grado 3 ó 4 notificada con mayor frecuencia en pacientes con mieloma mśltiple en recaķda/refractario fue la neutropenia, seguido de anemia y trombocitopenia. Se debe monitorizar a los pacientes en busca de posibles reacciones adversas hematológicas, especialmente neutropenia. Se debe advertir a los pacientes que informen rįpidamente acerca de los episodios febriles que presenten. Los médicos deben estar atentos a los signos de hemorragia en los pacientes, incluyendo epistaxis, especialmente en el caso de medicación concomitante conocida por aumentar el riesgo de sangrado (ver Efectos colaterales). Debe efectuarse a los pacientes un hemograma completo semanal en el momento basal, durante las primeras 8 semanas y después mensualmente. Puede ser necesaria una modificación de la dosis. Los pacientes pueden requerir el uso de hemoderivados y/o factores de crecimiento. Eventos tromboembólicos: Se han observado eventos tromboembólicos venosos (predominantemente trombosis venosa profunda y embolia pulmonar) y eventos trombóticos arteriales en pacientes tratados con pomalidomida en combinación con dexametasona. Los pacientes con factores de riesgo conocidos de tromboembolismo, incluida una trombosis previa, deben estar estrechamente monitorizados. Se deben tomar medidas para intentar minimizar todos los factores de riesgo modificables (p. ej. tabaquismo, hipertensión e hiperlipidemia). Se aconseja a médicos y pacientes que estén atentos a los signos y sķntomas de tromboembolismo. Se debe advertir a los pacientes que soliciten atención médica si presentan sķntomas como respiración entrecortada, dolor torįcico o edema de las extremidades. Es recomendable el uso de terapia anticoagulante (si no estį contraindicada), como el įcido acetilsalicķlico, warfarina, heparina o clopidogrel, especialmente en pacientes con factores de riesgo trombótico adicionales. Después de una cuidadosa evaluación de los factores de riesgo subyacentes del paciente individual, se debe tomar una decisión respecto al uso de medidas profilįcticas. En los estudios clķnicos los pacientes recibieron įcido aceltilsalicķlico profilįctico o terapia antitrombótica alternativa. El uso de agentes eritropoyéticos conlleva un riesgo de eventos trombóticos incluyendo tromboembolismo. Por lo tanto, deben emplearse con precaución los agentes eritropoyéticos asķ como otros agentes que puedan aumentar el riesgo de eventos tromboembólicos. Neuropatķa periférica: Se excluyó de los estudios clķnicos con pomalidomida a los pacientes con neuropatķa periférica en curso de Grado ³ 2. Se deben adoptar las precauciones adecuadas al considerar el tratamiento de estos pacientes con pomalidomida. Disfunción cardķaca significativa: Se excluyó de los estudios clķnicos con pomalidomida a los pacientes con una disfunción cardķaca significativa (insuficiencia cardķaca congestiva [Clase III o IV de la NY Heart Association]; infarto de miocardio dentro de los 12 meses desde el inicio del estudio; angina de pecho inestable o mal controlada). Se han notificado acontecimientos de insuficiencia cardķaca, que incluyen insuficiencia cardķaca congestiva y edema pulmonar (ver Efectos colaterales), especialmente en pacientes con enfermedad cardķaca preexistente o factores de riesgo cardķacos. Se deben adoptar las precauciones adecuadas al considerar el tratamiento de estos pacientes con pomalidomida, incluido el control periódico para detectar la presencia de signos y sķntomas de insuficiencia cardķaca. Sķndrome de lisis tumoral: Puede producirse un sķndrome de lisis tumoral. Los pacientes con mayor riesgo de sufrir un sķndrome de lisis tumoral son aquéllos que presentan una carga tumoral elevada antes del tratamiento. Se debe monitorizar estrechamente a estos pacientes y se deben adoptar las precauciones adecuadas. Segundas neoplasias malignas primarias: Se han notificado segundas neoplasias malignas primarias, como cįncer de piel no melanoma, en pacientes en tratamiento con pomalidomida (ver Efectos colaterales). Los médicos deben evaluar cuidadosamente a los pacientes antes y durante el tratamiento, utilizando pruebas estįndar de detección de cįncer por si aparecieran segundas neoplasias malignas primarias e instaurar el tratamiento indicado. Reacciones alérgicas:Se han notificado angioedema y reacciones dermatológicas graves (ver Efectos colaterales). Se excluyó de los estudios clķnicos a los pacientes con antecedentes de reacciones alérgicas graves asociadas a talidomida o lenalidomida. Estos pacientes pueden presentar un mayor riesgo de reacciones de hipersensibilidad y no deben tomar pomalidomida. Se debe considerar la interrupción o suspensión de pomalidomida si se presenta exantema de Grado 2 ó 3. Se debe suspender definitivamente el tratamiento con pomalidomida si se presenta angioedema, exantema de Grado 4 y exantema ampolloso o exfoliativo. Mareo y confusión: Se han notificado mareo y estados de confusión con pomalidomida. Los pacientes deben evitar las situaciones en que el mareo o la confusión puedan representar un problema y no tomar otros medicamentos que puedan causar mareo o confusión sin solicitar antes consejo médico. Enfermedad pulmonar intersticial (EPI): Se han observado EPI y acontecimientos asociados que incluyen casos de neumonitis con pomalidomida. Se debe realizar una evaluación cuidadosa de los pacientes que presenten un inicio repentino o empeoramiento inexplicable de los sķntomas pulmonares para descartar la EPI. Se debe interrumpir la administración de pomalidomida durante la investigación de estos sķntomas y, si se confirma la EPI, debe iniciarse un tratamiento adecuado. Śnicamente se debe reanudar pomalidomida después de una evaluación exhaustiva de los beneficios y los riesgos. Trastornos hepįticos: Se han observado concentraciones notablemente elevadas de alanina aminotransferasa y bilirrubina en los pacientes tratados con pomalidomida (ver Efectos colaterales). Se han notificado también casos de hepatitis que provocaron la suspensión de pomalidomida. Se recomienda controlar periódicamente la función hepįtica durante los primeros 6 meses de tratamiento con pomalidomida y, posteriormente, cuando esté clķnicamente indicado. Infecciones: Se han notificado rara vez casos de reactivación de la hepatitis B en pacientes en tratamiento con pomalidomida combinado con dexametasona que habķan sido previamente infectados por el virus de la hepatitis B (VHB). Algunos de estos casos han evolucionado a fallo hepįtico agudo, dando lugar a la suspensión de pomalidomida. Se debe determinar el estado del virus de la hepatitis B antes de iniciar el tratamiento con pomalidomida. Se recomienda que los pacientes que den un resultado positivo en la prueba de infección por VHB se pongan en contacto con un médico con experiencia en el tratamiento de la hepatitis B. Se debe tener precaución cuando se administre pomalidomida en combinación con dexametasona en pacientes previamente infectados por el VHB, incluidos los pacientes anti-Bc positivos pero con HBsAg negativos. Se debe monitorizar estrechamente a estos pacientes para detectar signos y sķntomas de infección activa por el VHB durante todo el tratamiento.
|
|
Precauciones:
Efectos sobre la capacidad para conducir y utilizar mįquinas: La influencia de Pomalist® sobre la capacidad para conducir y utilizar mįquinas es pequeńa o moderada. Se han notificado casos de fatiga, disminución del nivel de conciencia, confusión y mareo relacionados con el uso de pomalidomida. Si notan estos efectos, se debe advertir a los pacientes de que no deben conducir automóviles, utilizar mįquinas o realizar cualquier actividad peligrosa durante su tratamiento con pomalidomida. Fertilidad, embarazo y lactancia: Mujeres con capacidad de gestación/anticonceptivos en varones y mujeres: Las mujeres con capacidad de gestación deben utilizar dos (2) métodos anticonceptivos efectivos. Si una mujer tratada con pomalidomida se queda embarazada, se debe suspender el tratamiento y derivar a la paciente a un médico especialista o con experiencia en teratologķa, para su evaluación y asesoramiento. Si un paciente varón toma pomalidomida y su pareja se queda embarazada, se recomienda derivar a la mujer a un médico especialista o con experiencia en teratologķa, para su evaluación y asesoramiento. Pomalidomida estį presente en el semen humano. Como medida de precaución, todos los pacientes varones que tomen pomalidomida deben usar preservativos durante todo el tratamiento, durante la interrupción de la administración y hasta 7 dķas después del final del tratamiento, si su pareja estį embarazada o tiene capacidad de gestación y no estį usando ningśn método anticonceptivo. Embarazo: Se espera un efecto teratogénico de pomalidomida en humanos. Pomalidomida estį contraindicada durante el embarazo y en mujeres con capacidad de gestación, a menos que se cumplan todas las condiciones para la prevención del embarazo. Lactancia: Se desconoce si pomalidomida se excreta en la leche materna. Se detectó la presencia de pomalidomida en la leche de ratas que estaban siendo amamantadas tras administrar el medicamento a la madre. Debido a las posibles reacciones adversas en lactantes asociadas a pomalidomida, se debe decidir si es necesario suspender la lactancia o suspender el tratamiento tras considerar la importancia del tratamiento para la madre. Fertilidad: Se sabe que pomalidomida tiene un efecto negativo sobre la fertilidad y que es teratogénica en los animales. Tras su administración a conejas embarazadas, pomalidomida atravesó la placenta y se detectó en la sangre fetal. Carcinogénesis, mutagénesis y fertilidad: Datos preclķnicos sobre seguridad: Estudios de toxicidad de dosis repetidas: La administración crónica de pomalidomida en ratas en dosis de 50, 250 y 1000 mg/kg/dķa durante 6 meses fue bien tolerada. No se detectó ningśn efecto adverso hasta los 1000 mg/kg/dķa (una tasa de exposición 175 veces mįs elevada que la dosis clķnica de 4 mg). Se evaluó pomalidomida en monos en estudios de dosis repetidas de hasta 9 meses de duración. En estos estudios, los monos mostraron una mayor sensibilidad a los efectos de pomalidomida que las ratas. Las toxicidades primarias observadas en monos estuvieron relacionadas con los sistemas hematopoyético/linforreticular. En el estudio de 9 meses en monos con dosis de 0.05, 0.1 y 1 mg/kg/dķa se observó morbilidad y eutanasia temprana de 6 animales a dosis de 1 mg/kg/dķa que fueron atribuidas a los efectos inmunosupresores (infección por estafilococos, reducción de los linfocitos en sangre periférica, inflamación crónica del intestino grueso, reducción histológica de los linfocitos e hipocelularidad de la médula ósea) a exposiciones elevadas de pomalidomida (15 veces la tasa de exposición comparada con una dosis clķnica de 4 mg). Dichos efectos inmunosupresores provocaron la eutanasia temprana de 4 monos debido a su mal estado de salud (heces lķquidas, inapetencia, ingesta de alimentos reducida y pérdida de peso); la evaluación histopatológica de estos animales demostró inflamación crónica del intestino grueso y atrofia vellosa del intestino delgado. Se observó infección por estafilococos en 4 monos; 3 de éstos respondieron al tratamiento con antibióticos y 1 murió sin tratamiento. Ademįs, resultados consistentes con la leucemia mielógena aguda llevaron a la eutanasia de un mono; las observaciones clķnicas y la patologķa clķnica y/o alteraciones de la médula ósea observadas en este animal eran consistentes con inmunosupresión. La proliferación mķnima o leve en los conductos biliares con incrementos asociados de la ALP y de la GGT también se observaron a dosis de 1 mg/kg/dķa. La evaluación de los animales recuperados indicó que todos los resultados relacionados con el tratamiento eran reversibles a las 8 semanas del cese de la administración, excepto la proliferación de los conductos biliares intrahepįticos observada en 1 animal en el grupo de 1 mg/kg/dķa. El nivel sin efecto adverso observado (NOAEL) fue de 0.1 mg/kg/dķa (una tasa de exposición relativa de 0.5 veces comparada con la dosis clķnica de 4 mg). Genotoxicidad/carcinogenicidad: Pomalidomida no resultó mutagénica en los ensayos de mutaciones bacterianas y de los mamķferos, y no indujo aberraciones cromosómicas en los linfocitos de sangre periférica en humanos asķ como tampoco a la formación de micronścleos en eritrocitos policromįticos en la médula ósea de ratas a las que les fueron administradas dosis de hasta 2000 mg/kg/dķa. No se han realizado estudios de carcinogenicidad. Fertilidad y desarrollo embrionario temprano: En 1 estudio de fertilidad y desarrollo embrionario temprano en ratas, se administró pomalidomida a los machos y las hembras a dosis de 25, 250 y 1000 mg/kg/dķa. El examen uterino en el dķa de gestación 13 mostró una reducción de la cantidad media de embriones viables y un aumento en la pérdida post-implantación con todos los niveles de dosis. Por consiguiente, el NOAEL en estos eventos observados fue <25 mg/kg/dķa (con un AUC24h de 39960 ng · h/ml (nanogramo por hora/mililitros) para la dosis mįs baja evaluada y una tasa de exposición 99 veces relativa a la dosis clķnica de 4 mg). Cuando los machos tratados en este estudio fueron apareados con hembras no tratadas, todos los parįmetros uterinos fueron comparables a los controles. Segśn estos resultados, los efectos observados fueron atribuidos al tratamiento de las hembras. Desarrollo embriofetal: Pomalidomida resultó ser teratógena en ratas y conejos cuando se administró durante el perķodo de mayor organogénesis. En el estudio de toxicidad sobre el desarrollo embriofetal de la rata, se observaron malformaciones relacionadas con la ausencia de vejiga urinaria, ausencia de la glįndula tiroidea, asķ como la fusión y la desalineación de los elementos vertebrales torįcicos y lumbares (arcos centrales y/o neurales) a todos los niveles de dosis (25, 50 y 1000 mg/kg/dķa). En este estudio no se observó toxicidad materna. por ello, el NOAEL materno fue de 1000 mg/kg/dķa, y el NOAEL para la toxicidad de desarrollo fue <25 mg/kg/dķa (con un AUC24h de 34340 ng.h/ml en el dķa de gestación 17 para la dosis mįs baja evaluada y la tasa de exposición fue de 85 veces comparada con la dosis clķnica de 4 mg). En conejos, pomalidomida a dosis entre los 10 y 250 mg/kg/dķa produjo malformaciones en el desarrollo embriofetal. Se observaron aumentos de las anomalķas cardķacas a todas las dosis con aumentos significativos a 250 mg/kg/dķa. A dosis de entre 100 y 250 mg/kg/dķa se registró un ligero aumento de la pérdida post-implantación y un ligero descenso en el peso del feto. A dosis de 250 mg/kg/dķa, las malformaciones fetales incluyeron anomalķas en las extremidades (extremidades anteriores y posteriores dobladas y/o giradas, ausencia de dķgito o dķgito libre) y malformaciones esqueléticas asociadas (metacarpiano no osificado, metacarpiano y falange no alineados, ausencia de dķgito, falange no osificada y tibia corta no osificada o doblada); dilatación moderada de los ventrķculos laterales del cerebro; ubicación anormal de la arteria subclavia derecha; ausencia de los lóbulos intermedios pulmonares; par de rińones desplazados hacia abajo; morfologķa hepįtica alterada; ausencia de osificación de la pelvis u osificación incompleta; aumento medio de las costillas torįcicas supernumerarias y reducción media de los tarsales osificados. Ademįs, se observó una ligera reducción en el incremento del peso materno, una reducción significativa de los triglicéridos, y una reducción significativa del peso absoluto y relativo del bazo a dosis de entre 100 y 250 mg/kg/dķa. El NOAEL materno fue de 10 mg/kg/dķa y el NOAEL del desarrollo fue de <10 mg/kg/dķa (con un AUC24h de 418 ng · h/ml en el dķa de gestación 19 para la dosis mįs baja evaluada, similar a la obtenida con una dosis clķnica de 4 mg).
|
|
Interacciones Medicamentosas:
Efecto de Pomalyst®sobre otros medicamentos: No se espera que pomalidomida pueda causar interacciones medicamentosas farmacocinéticas clķnicamente relevantes debido a la inhibición o inducción de la isoenzima P450, o inhibición de transportadores cuando se administra de forma concomitante con sustratos de estas enzimas o transportadores. No se ha evaluado clķnicamente el potencial de estas interacciones medicamentosas, incluyendo el posible impacto de pomalidomida en la farmacocinética de los anticonceptivos orales combinados. Efecto de otros medicamentos sobre Pomalyst®: Pomalidomida se metaboliza parcialmente por CYP1A2 y CYP3A4/5. También es un sustrato de la glicoproteķna P. La administración concomitante de pomalidomida con ketoconazol, inhibidor potente del CYP3A4/5 y de la Gp-P, o con el inductor potente del CYP3A4/5, carbamazepina, no demostró ningśn efecto clķnicamente relevante a la exposición a pomalidomida. La administración concomitante de pomalidomida con el inhibidor potente del CYP1A2 fluvoxamina en presencia de ketoconazol, incrementó la exposición media a pomalidomida en un 107%, con un intervalo de confianza del 90% [del 91% al 124%], frente a pomalidomida mįs ketoconazol. En un segundo estudio realizado para evaluar la contribución a los cambios del metabolismo de un inhibidor del CYP1A2 solo, la administración conjunta de fluvoxamina sola con pomalidomida aumentó la exposición media a pomalidomida en un 125% con un intervalo de confianza del 90% [del 98% al 157%] frente a pomalidomida administrada en monoterapia. Si se administran inhibidores potentes del CYP1A2 (p. ej., ciprofloxacino, enoxacino y fluvoxamina) de forma concomitante con pomalidomida, se debe reducir la dosis de pomalidomida en un 50%. Dexametasona: La administración concomitante de mśltiples dosis de hasta 4 mg de pomalidomida con dexametasona de 20 mg a 40 mg (un inductor leve a moderado de varias enzimas CYP, incluido el CYP3A) en pacientes con mieloma mśltiple no tuvo ningśn efecto sobre la farmacocinética de pomalidomida frente a pomalidomida administrada sola. Se desconoce el efecto de dexametasona sobre la warfarina. Se aconseja realizar una monitorización rigurosa de la concentración de warfarina durante el tratamiento.
|
|
Sobredosificación:
Sobredosis y tratamiento: Se han evaluado dosis de pomalidomida de hasta 50 mg en dosis śnica en voluntarios sanos y 10 mg en mśltiples dosis 1 vez al dķa en pacientes con mieloma mśltiple sin que se haya notificado ningśn caso de efecto adverso grave relacionado con sobredosis. Pomalidomida se eliminó mediante hemodiįlisis. En caso de sobredosis, se recomienda terapia de soporte. Ante la eventualidad de una sobredosificación, concurrir al hospital mįs cercano o comunicarse con laboratorios tecnofarma al teléfono 225949201 o a la dirección electrónica "www.tecnofarma.cl"
|
|
Conservación:
Conservar a temperatura no mayor a 30° C.
|
|
Presentaciones:
Cada envase contiene 21 cįpsulas.
|
|



{kind=link}
{kind=link}
{kind=link}
{kind=link}